Nilanjana Biswas1 Sangita Ghosh1 and Arijit Bag2,3
1Department of Chemistry, Sree Chaitanya College, Habra, West Bengal, India
2Department of Chemical Sciences, Indian Institute of Science Education and Research, Kolkata, West Bengal, India
3Department of Applied Sciences, Maulana Abul Kalam Azad University of Technology, Simhat, Haringhata, West Bengal, India
*Corresponding Author: Arijit Bag, Department of Chemical Sciences, Indian Institute of Science Education and Research and Department of Applied Sciences, Maulana Abul Kalam Azad University of Technology, Simhat, Haringhata, West Bengal, India.
Received: August 24, 2020; Published: February 27, 2021
Background: In spite of so many FDA approved HIV drugs, HIV infections and deaths are increasing across the glob. Thus, HIV research is still important and challenging. Finding of low cost highly effective mutation resistant HIV drug is the prime destination of HIV researchers. Small organic molecules may be cost effective if they provide desired drug activities in this regards.
Objective: Finding of small organic molecules with high HIV drug potency is the main target of present research. Since HIV-1 capsid assembly inhibitors (CA) are mutation resistant, we focused on finding of inhibitors of this class.
Methods: Few randomly selected small organic molecules are tested as HIV-1 CA inhibitors through molecular docking using Docking Server. Only effective compounds are used for further calculations to predict IC50, CC50, LogP. For these cal-culations, Density Functional theory (DFT), Quantum Computation Methodology (QCM), Relative IC50 methodology (RICM) and group contribution methodology for LogP calculation are used.
Results: HIV-1 CA inhibitory capacity of p-di-pyrrole benzene and its derivatives are found very good from the docking methods. It is observed that, among three compounds which are promising, o-ethyl p-di-pyrrole benzene has the highest inhibition constant as an HIV-1 CA inhibitor. Quantum computations of IC50, CC50 and LogP also supported the docking results. o-ethyl p-di-pyrrole benzene shows desirable activity as HIV-1 CA inhibitor.
Conclusion: Since o-ethyl p-di-pyrrole benzene is a small organic molecule it would be easily synthesizable and cost-effective. Thus, it may be a very good HIV drug. To exaggerate its prospect we have also studied and reported different important properties of these compounds like IC50, CC50, LogP, etc., which are required before pre-clinical trials. These results are also in favor of o-ethyl p-di-pyrrole benzene to be a drug in practice.
Keywords: HIV-1 Capsid A Inhibitor; IC50; CC50; LogP; DFT; p-di-Pyrrole Benzene
There are more than 25 FDA-approved HIV-1 drugs [1-4] for seven classes; nucleoside reverse transcriptase inhibitors (NRTIs), nucleotide reverse transcriptase inhibitors (NtRTIs), non-nucleoside reverse transcriptase inhibitors (NNRTIs), protease inhibitors (PIs), fusion inhibitors (FIs), co-receptor inhibitors (CRIs) and integrase inhibitors (INIs) [5] that target one of the three viral enzymes require for completing its (HIV virus) life cycle in the host body (human body) [6]. There are three key steps involved in viral attack; attachment, replication and assembling [6-9]. HIV virus after entering into the blood circulation system of human body, tries to get attached to CCR5 receptor [10] of CD4 cells [11], which is one of the two T-cell types lymphocytes of white blood cell. CD4 cells which control and direct the immune response and coordinate to other immune cells, act as a virus manufacturing machine after HIV infection. In spite of many avail-able drugs, lifelong therapy is required for sustained viral suppression. Due to very fast mutation of HIV-1 [12,13], drug resistance is acquired within very short period of time. Moreover, cross-resistance [14] within mechanistic classes underscoring the need to discover new class of HIV-1 inhibitor.
HIV-1 capsid (CA) protein plays essential roles in the viral replication process which is an attractive new therapeutic target for antiretroviral treatment [15]. During assembling and budding the proteolytic cleavage of 55-kDa Gag polyprotein yields CA protein which provides key protein- protein interactions among immature virion proteins to assemble into the cone- shaped central capsid that surrounds the viral RNA genome, reverse transcriptase (RT) and integrase (IN) [16]. CA mutations impair the assembly and inhibit viral replication. N-terminal domain of CA (CANTD) includes 1 to 146 residues [17] and C-terminal domain (CACTD) is composed of 151 to 231 residues [18]. These two highly helical domains are connected through a very short (only four residues) flexible linker. High-resolution structures of CA hexagons and pentagons are reported by Pornillos., et al. [19-21]. There are three independent inhibitor binding sites on CA; at the junction of helices 1, 2, 4 and 7, a conserved hydrophobic cleft within the CACTD four-helix bundle and between helix 4 and helix 7 of the CANTD [22]. CA binding inhibitors of several classes have been reported and providing evidence that CA may be a viable drug target. Christopher., et al. [23] studied two different classes of capsid inhibitors and found that mutations conferring resistance to both classes of compound had two different types of effects. Substitutions within the inhibitor binding site decrease the inhibitor binding affinity, but at the same time viral fitness also decreases. Thus, though due to mutation inhibitor binding probability decreases, viral replication processes do not enhance. This suggests that the development of drug resistance against capsid assembly inhibitors due to mutation is not possible for the HIV virus. Christopher., et al. also shown that due to mutation, stability of inhibitor-enzyme complex increases. Thus, possibility of the development of drug resistance against capsid assembly inhibitors is very less for those inhibitors whose protein binding free energies are highly negative.
In spite of enormous effort, highly efficient HIV Capdi-A inhibitor drug is not market available, though, different groups around the world are continuously working in this field and prospective results are reported in a rapid basis [24-26]. At present, sensational progress in this field is achieved, but, the unavailability of drugs is mostly due to the cost effectiveness of the product reported. Thus, we model very small organic molecules which should be synthetically cost effective and at the same time highly active. For this purpose we systematically study HIV Capsid-A inhibitor capacity of different isomers of di-pyrrole benzene and its alkyl and halide derivatives. For geometry optimization we use Density Functional Theory (DFT) calculation with the help of GAUSSIAN 09 package. Molecular docking method is used for binding affinity calculation using docking server.
The aim of this work is to find out small organic com-pounds which will be active as HIV-1 CA inhibitor. Thus, we started to compute the bio-activity of small organic com-pounds using molinspiration [27] which predict the bio-activity from quantitative structure-property relationships (QSPR) calculatio [28]. This program package works on-line. We have studied few such small molecules up to a maximum of three substitutions, systematically. Among nearly thirty odd samples, we have chosen only six samples for our docking study which showed better bio-activity among these compounds. After docking we have chosen only three compounds for Density Functional Theory (DFT) study which has IC50 in the micro molar rang. Only these compounds are reported here.
Methods used for different calculations Molecular Docking methodMolecular docking methods [29-31] are well known and used globally for protein ligand interaction study. Molecular docking uses a classical force field for energy calculation. By the energy minimization search we determine the most probable binding site of a protein. The binding constant is also countable from docking. In the present study, Docking Server [32] is used for the docking calculation. This also works on-line. We used only the free accessed version. Different docking parameters which are used for our calculation is presented in table 1.
Parameters |
Value |
tstep |
0.2 |
qstep |
5.0 |
dstep |
5.0 |
rmstol |
2.0 |
ga-pop-size |
150 |
ga-num-evals |
250000 |
ga-num-generations |
540000 |
ga-run |
10 |
Table 3: Screening of genotypic correlation with grain yield into direct (bold) and indirect effect of outcrossing and yield granting characters in restorer line of rice. * represent significant at 5% level of significance, ** represent significant at 1% level of significance. Residual effect, R= 0.5952.
Gasteiger partial charges were added to the ligand atoms. Non-polar hydrogen atoms were merged, and rotated bonds were defined. Docking calculations were carried out on di- py-ben protein model. Essential hydrogen atoms, Kollman united atom type charges, and solvation parameters were added with the aid of AutoDock tools. Affinity (grid) maps of xx Å grid points and 0.375 Å spacing were generated using the Auto grid program. AutoDock parameter set- and distance-dependent dielectric functions were used in the calculation of the van der Waals and the electrostatic terms, respectively. Docking simulations were performed using the Lamarckian genetic algorithm (LGA) and the Solis & Wets local search method. Initial position, orientation, and torsions of the ligand molecules were set randomly. All rotated torsions were released during docking. Each docking experiment was derived from 10 different runs that were set to terminate after a maximum of 250000 energy evaluations. The population size was set to 150. During the search, a translational step of 0.2 Å, and quaternion and torsion steps of 5 were applied.
Quantum IC50 Computation Method (QICM)For IC50 computation of our modeled compounds using DFT, we pursue two different methodologies, Quantum IC50 Computation Method (QICM) and Relative IC50 Computation Method (RICM) which are recently developed by Bag [33], Bag and Ghorai [34]. QICM is based on the basic en-zyme ligand reaction kinetics proposed by L. Michaelis and M. Menten. From Michaelis-Menten equation we get

Where, Ki and Ks are binding constant of inhibitor and substrate respectively. [S] is the substrate concentration. If we replace binding constants by binding free energy we shall get.

Where,

ΔG0i and ΔG0s are the standard Gibbs free energy change for inhibitor and substrate binding with the enzyme, respectively. Thus, Θ is a process specific constant. We have to evaluate it once for a particular process. To reduce the computation cost, another modification of this method was done and shown effective, accurate and applicable. In this method, (ΔG0i)/RT is modeled as

Here, B is a constant which depends on temperature of the enzymatic process and other external conditions which influence the process. µ and µ* are dipole moment and hydrophobicity of the inhibitor, respectively. ω is the reactivity index [35] of the inhibitor.
Relative IC50 computation method (RICM)To reduce the computation cost and increase the implementation territory, an alternative methodology (RICM) to QICM is developed by Bag very recently which is used for the present calculation. Thus, RICM is described in brief here. In RICM, a relative IC50 of an inhibitor is calculated using the IC50 value of a similar inhibitor for a particular process. Thus, the results depend on the choice of the reference compound. But, this methodology is very simple and able to produce very good results as accurate as the QCM method [33]. The equation for computation of IC50 using this method is
 DFT Computational Method
DFT Computational Method
In DFT method [36-38], the energy of a system is described in terms of the electron density in space. The electron density is described by a functional. There are several functional developed so far and still developing. Few functional which are frequently used by the most computational chemistry users, are B3LYP [39], B3PW91 [40], etc. Several market available software included DFT calculations for different purposes. Gaussian [41], GAMESS [42], Q-chem [43], Quantum Expresso [44], VASP [45], NWChem [46], etc. are the leading software in this regards. In general, first, the geometry of a compound is optimized unless exact geometry is available from the experiment. With the optimized geometry different properties of a molecule is computed. To accomplish our present study, we first optimized the structures of our compounds using Gaussian 09 package. These optimized geometries are used to compute dipole moment and the orbital energies which are required to calculate reactivity descriptors.
All geometry optimizations of our used compounds reported here are done following standard procedures taking convergence threshold as 0.000015 Hartree/Bohr. Calculations are performed within the density functional theory (DFT) approximation taking B3LYP functional.
Three compounds which are found as active HIV-1 CA inhibitors after molecular docking calculations are 1-[4-(1H- pyrrol-1-yl) phenyl]-1H-pyrrole (ligand-1), 1,4-dicyclopentyl- 2-methylbenzene (ligand-2) and 1,4-dicy-clopentyl-2- ethylbenzene (ligand-3). We first chose pyrrole as substitution because in guanine, pyrrole structure exists. We computed binding affinity of this ligand (from here and onwards, it would be termed as ligand-1) with p24 HIV Capsid-A protein (PBD ID is 1e6j). Then we systematically substitute methyl, ethyl, iodine, chlorine and hydroxyl group to our ligand to search for improvement in binding affinity. Unfortunately, no significant improvement was observed. Thus, for getting better activity we used cyclopentyl functional group as main substitution instead of pyrrole. We observed good activity with this substitution. This was followed by methyl and ethyl substitution at the benzene ring ortho to a cyclopentyl group. Docking and DFT results of these two compounds are reported here.
Docking results of Ligand-1 with HIV-1 CA proteinDocked structure of ligand-1 with HIV-1 capsid-A protein is presented at figure 1. It is observed that as our ligand is small it easily goes to the binding site. But it is too small to tightly fit to the binding cavity. As a result, binding energy is very small, only -4.9 kcal/mol. There is only one hydrogen bonding formation is observed which is presented in figure 2. This hydrogen bonding is made by a glutamate residue which is 155th residue. All other interactions are Van der Waals interaction. Though its binding energy is small, its binding frequency is very high, as high as 30.0%. It is because its size is very small and its charge density is very high. It has three interaction zones which is shown in HB plot presented in figure 3. Estimated inhibition constant for this ligand is 250 μ mole.

Figure 1: Docked structure of ligand-1 with p24 HIV protein.

Figure 2: Interacting residues of p24 HIV protein when docked with ligand-1.
Docking results of Ligand-2 with HIV-1 CA proteinDocked structure of ligand-2 is presented in figure 4 and interaction plot is presented in figure 5. Binding affinity of ligand-2 is -5.13 kcal/mol and binding frequency is 50.0% which is very high. Thus, ligand-2 is an improved inhibitor. It is observed that ligand-2 is attached to one side of the cavity. Thus, its binding frequency is very high. Inhibition constant of this ligand is 174.9 μ mole.
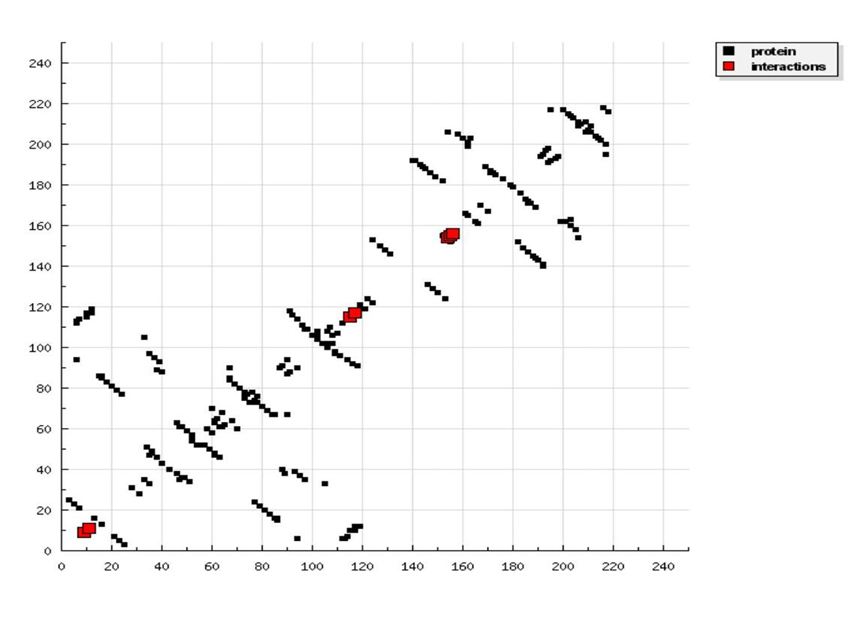
Figure 3: HB plot of ligand-1 with p24 HIV protein.
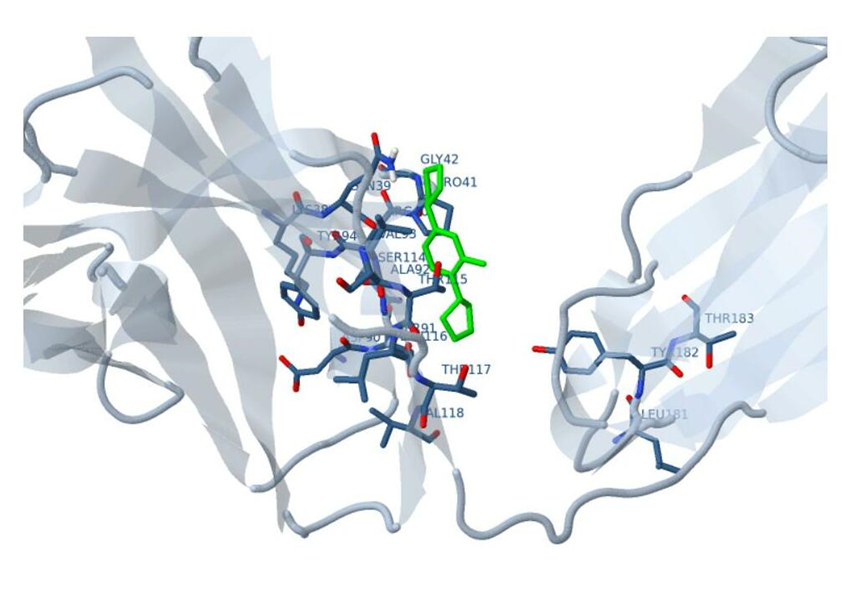
Figure 4: Docked structure of ligand-2 with p24 HIV protein.
Docking results of Ligand-3 with HIV-1 CA proteinWe observed that ligand-2 is an improved ligand as a HIV-1 capsid inhibitor compare to ligand-1. Thus, for further improvement we substitute with ethyl at the ortho position of benzene ring (ligand-3). Docked structure of ligand-3 is presented in figure 6 and interaction is plotted in figure 7. Binding affinity of ligand-3 is -6.17 kcal/mol and binding frequency is 20.0% which is not high as its methyl counter part but it is acceptable. Its binding frequency is less due to its larger size. It is observed that ligand-3 is attached to the centre of the cavity. Inhibition constant of ligand-3 is only 29.78 μ mole, which is excellent and comparable to market available drugs. Thus, this ligand may be a new drug for HIV-1 capsid-A inhibitor. But for that, this has to pass biological tests.
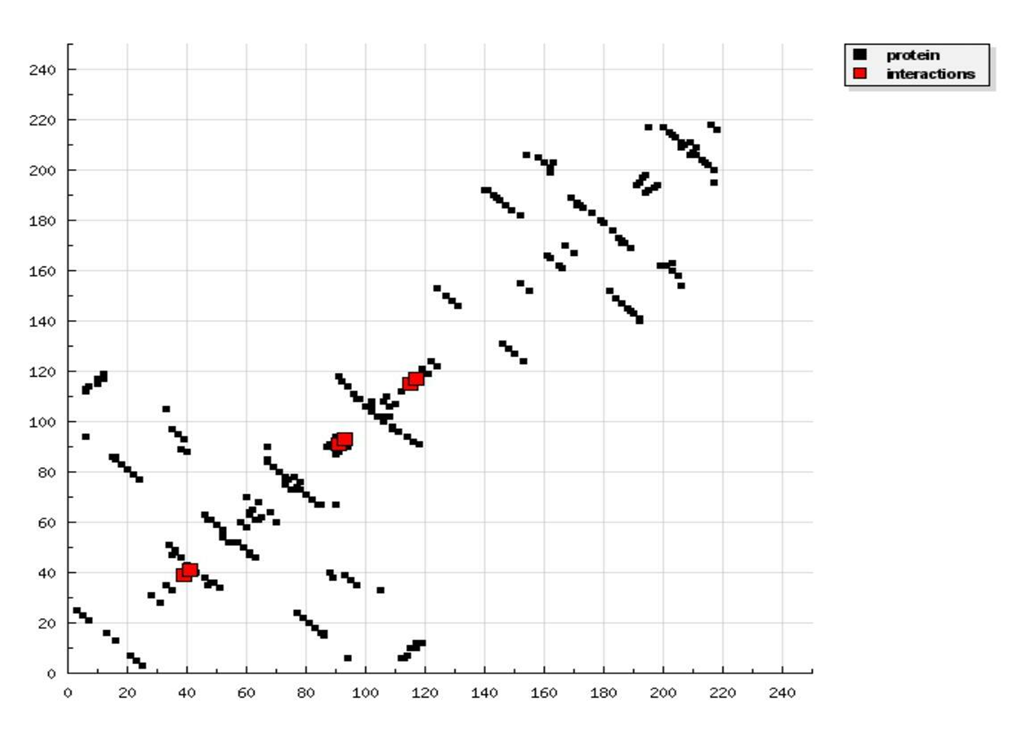
Figure 5: HB plot of ligand-2 with p24 HIV protein.
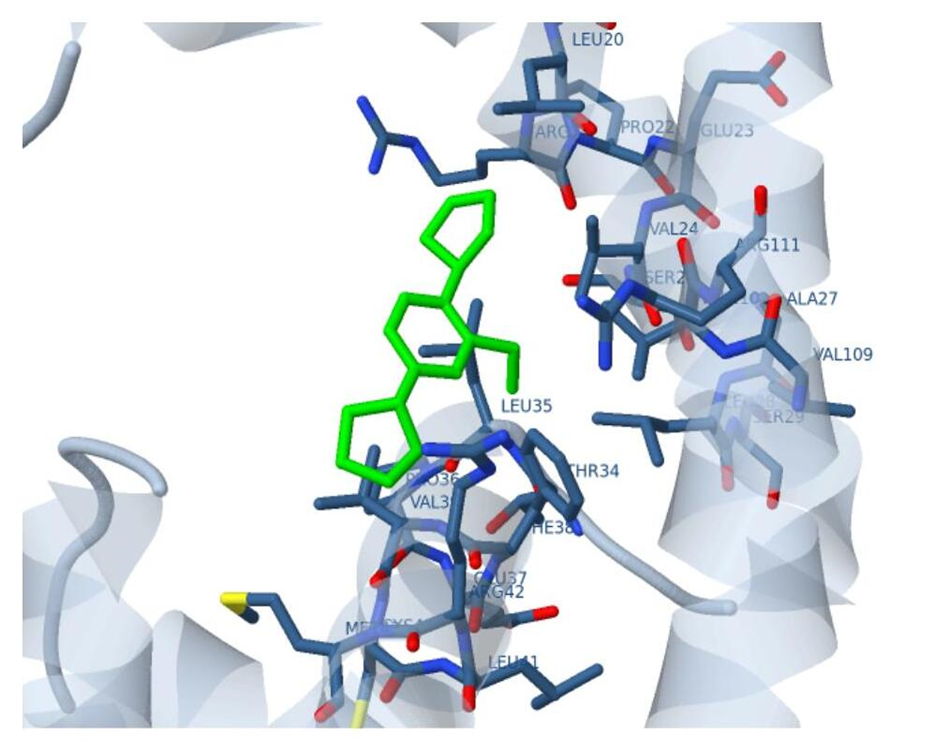
Figure 6: Docked structure of ligand-3 with p24 HIV protein.
DFT results: IC50, CC50 and LogPFrom the docking study we observed that three p-di-pyrrole benzene derivatives show good activity as HIV-1 Capsid inhibitor. Thus, we compute IC50, CC50 and LogP of these compounds. For this purpose, we optimized these compounds using Gaussian 09. The optimized geometry of o-ethyl p-di-pyrrole benzene is presented in figure 8. We used two different methods of IC50 computation, quantum computation method (QICM) developed by Bag and Ghorai [34] and relative IC50 computation method (RICM) proposed by Bag [33]. LogP is computed using on-line platform of molinspiration which has very good accuracy [47]. Toxicity of these compounds (CC50), are also computed using relative toxicity method prescribed by Bag and Ghorai [35]. Computed results are presented in table 2.
| Compounds | Dipole moment (D) | Log P | IC50a (mM) | CC50b (mM) |
|---|---|---|---|---|
Ligand-1 |
0.001 |
2.25 |
96 |
65.8 |
Ligand-2 |
0.598 |
2.67 |
29.3 |
110 |
Ligand-3 |
0.838 |
3.25 |
5.3 |
132 |
Table 2: Bio-activity properties of different ligands.

Figure 7: HB plot of ligand-3 with p24 HIV protein.
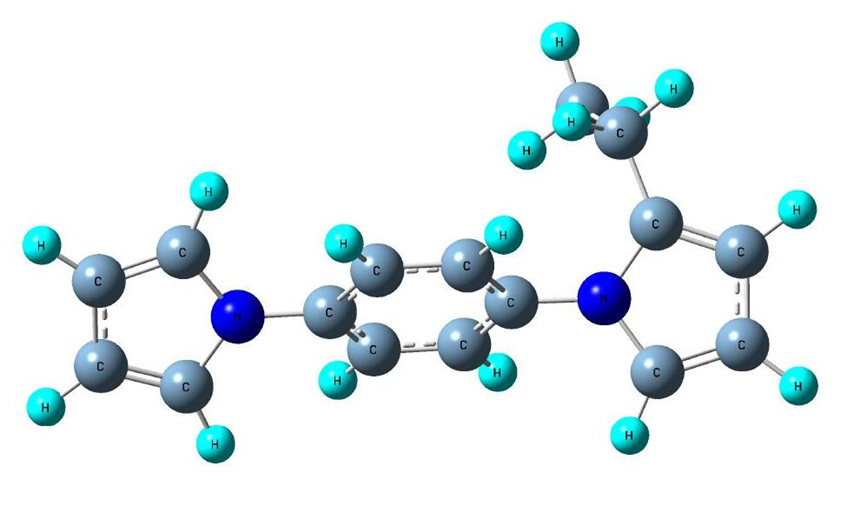
Figure 8: Optimized geometry of Ligand-3.
From the present study, it is observed that molecules which have high dipole moments exhibit higher LogP and CC50 values but lower IC50 values. It is known that in general, molecules having high hydrophobicity are less toxic. Present study is correlated with this fact. The LogP value of Ligand-3 (3.25) is the highest among the three ligands reported here. The CC50 value of this ligand (132) is the largest. It is also found that high dipole moment and LogP values are associated with low IC50 values which means they are highly active HIV-1 CA inhibitors. These results are obvious. But the most important observation of this study is that to improve the LogP and reduce the IC50 value of a compound requires the addition of hydrocarbon chain with proper increase of dipole moment.
Comparison of different theoretical resultsIt is very important to compare the IC50 values obtained from different theoretical models to endorse a model for future studies. Thus, we have presented the computed IC50 values obtained from different theoretical tools in table 3. From the presented values of IC50, it is observed that the order of RICM and QICM IC50 values obtained from RICM and QICM, are same as we obtained from the docking results though the values are not same. Quantum computed IC50 values are lower compared to the respective docking IC50 values. Bag [33] has shown that quantum computation of IC50 produces better results compared to the experimented IC50 than the molecular docking method of IC50 due to the inclusion of chemical behaviour of the inhibitors. Though the quantum computation method [34] is very accurate and we also did the molecular docking based IC50 computation of our compounds, still, we further compute IC50 of these compounds using the relative toxicity method [33] which are also presented in Table - 3, for the test of the newly reported approach of IC50 computation. For this calculation Cyclophiline A is used because, it is the substrate of this process. These IC50 values are higher compared to both molecular docking values and quantum computed values. But importantly, the order of IC50 values is same. It is mentioned that substrate based computation of relative IC50 is not accurate as it is for similar compound based computation [33]. In spite of this fact, we report these values to show that these compounds are really capable of inhibiting the substrate, Cyclophiline A.
| Compounds | Relative IC50 (mM) | Quantum IC50 (mM) | Docking IC50 (mM) |
|---|---|---|---|
p-di-pyrrole benzene |
521 |
96 |
250 |
o-methyle p-di-pyrrole benzene |
279 |
29.3 |
175 |
o-ethyl p-di-pyrrole benzene |
224 |
5.3 |
29.8 |
Table 3: Bio-activity properties of different ligands.
Comparison with previous resultsAt present, state of the art HIV research focused on CA inhibition as this class of drugs are mutation registrant drugs. Presently a handful of HIV-1 CA inhibitors are reported [24-26,49]. Different ligands have preferences for different binding sites also. Detailed theoretical and experimental studied have been carried with these compounds. Their binding affinity range from μM to nM. In another research work [50], we found that Ru5(CO)14(μ-H)(μ-S(CH2)2COO-)Na+ has the HIV-1 CA inhibition activity at the picomolar level. Still, we are promoting our present model ligands as HIV-1 CA inhibitor which has the activity at the micro molar level because, these are very common chemicals, very small in size, high binding frequency as well. Thus, these compounds would be cost-effective HIV drugs. In this regards, present work is very important and will be fruitful for future research.
In this research work small organic molecules are modelled and tested for HIV-1 Capsid-A inhibitor capacity. It is observed that 1,4-dicyclopentyl-2- ethylbenzene has very good inhibition constant (5.3 μM computed using quantum computation method). Though, several inhibitors of the same class with better activity is reported, this com-pound has the privilege in terms of production cost as it is a very small organic compound and shows very less toxicity with good hydrophobicity. Thus, this compound could be pursued for in-vitro test.
Citation: Arijit Bag.,et al.. “p-di-pyrrole Benzene Derivatives - A New Class of Highly Active HIV-1 CA Inhibitors". Acta Scientific Paediatrics 5.3 (2021): 92-100.
Copyright: © 2021 Arijit Bag., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.















 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
