Saeful Amin*, Anna Yuliana, Ira Rahmiyani and Fasya Ammatul Hawa
Department of Pharmacy, Bakti Tunas Husada University, Indonesia
*Corresponding Author: Saeful Amin, Department of Pharmacy, Bakti Tunas Husada University, Indonesia.
Received: July 26, 2024; Published: August 04, 2024
Citation: Saeful Amin., et al. “Virtual Screening of Secondary Metabolites from Abuta (Cissampelos pareira Linn) as Potential COVID-19 Inhibitors: A Computational Approach to Drug Discovery". Acta Scientific Pharmaceutical Sciences 8.9 (2024):03-21.
Following the declaration of COVID-19 as a pandemic, researchers have extensively investigated plant-derived compounds for their potential in treating and preventing the disease. Abuta (Cissampelos pareira Linn) has long been used in Ayurvedic by the people of India to treat various health problems. The secondary metabolite of abuta (Cissampelos pareira Linn) is known to have antiviral activity. This research employs computational methods to evaluate the potential of abuta (Cissampelos pareira Linn) secondary metabolites as inhibitors of SARS-CoV-2. Molecular docking analysis revealed that among abuta (Cissampelos pareira Linn) secondary metabolites, Isochondodendrine showed the highest affinity for the 5R7Y receptor with a score of -94.60, Cissampaline for 7JKV with -94.87, Kaempferol-3-O-β-D-Glucopyranoside for 7TLL with -99.03, and Cissampline for 7VH8 with -99.03. A compound that has good docking values at 5R7Y, 7JKV, 7TLL, and 7VH8 receptors; meets Lipinski’s rule of five, and has good absorption, distribution, and excretion values, and is not hepatotoxic is Cissampline. The interaction between the compound and the four receptors was also stable at the time of molecular dynamics simulation for 10 nanoseconds. Thus, Cissampeline can be said have potential as an anti-COVID-19.
Keywords: Abuta; Cissampelos pareira; COVID-19; In Silico Studies; Molecular Docking; Molecular Dynamics
The COVID-19 pandemic has spurred extensive research into plant-derived compounds for their potential to treat and prevent the disease. One plant of interest is Abuta (Cissampelos pareira Linn), traditionally used in Indian Ayurvedic medicine. This species has a long history in folk remedies, being employed to address various ailments including diarrhea, asthma, kidney stones, liver problems, abdominal pain, coughing, and fever [1].
According to in vitro studies, various chemicals found in abuta (Cissampelos pareira Linn) as well as extracts from all parts of the abuta plant can suppress SARS-CoV-2 by around 60% [2]. Therefore, this study aims to conduct an in silico study on the potential of secondary metabolites found in abuta (Cissampelos pareira Linn) plants as anti-COVID-19 compounds. The abuta plant (Cissampelos pareira Linn) was chosen for this study because the entire plant extract (Cissampelos pareira Linn) contains antiviral properties with an inhibitory concentration of 60% [3]. The method used is a study of molecular docking and molecular dynamics of compounds contained in the abuta plant (Cissampelos pareira Linn) against the COVID-19 receptor. This molecular anchoring study has the advantage of being a simple method for discovering new ligand. This method provides access to more compounds and is less expensive than laboratory-based methods [4]. For drug designers, in silico studies can be beneficial since they can be used to estimate the potential of a compound’s biological action against a disease so that it is not useless to synthesize it [5].
The main protease-type COVID-19 receptor was utilized in this study. Specifically, the receptors examined were 5R7Y, 7JKV, 7TLL, and 7VH8. The study compared these receptors against four antiviral drugs: Favipiravir, Molnupiravir, Nirmatrelvir, and Remdesivir. Remdesivir is an antiviral approved by the Food and Drug Administration for the treatment of COVID-19 [6]. Based on systematic reviews and meta-analyses, favipiravir can cause viral clearance in 7 days and contribute to clinical improvement in 14 days [7]. Molnupiravir is the newest type of COVID-19 drug in development, having completed phase 2 trials [8]. Nirmatrelvir is an oral antiviral that is given as a booster with Ritonavir packed as an oral medication made by Pfizer, known as Paxlovid [9].
The equipment used in this study include computer hardware and software. The hardware is a computer with Windows 10 specifications: 4.00 GB of memory (RAM). system type 64-bit operating system, x64-based processor, and the software used is PDB (Protein Data Bank), MOE.2009, MarvinSketch 21.18, PubChem, PLANTS, YASARA (serial number: 875143296), SwissDock, pkCSM, Chemical.AI, and AMBER.
The material used in this study was the PDB file identified by the main protease (MPro) coronavirus receptor, namely 5R7Y, 7JKV, 7VH8, and 7TLL with comparative drug compounds, namely Favipiravir, Remdesivir, Molnupiravir, and Nirmatrelvir which were downloaded from https://www. rcsb.org/ and secondary metabolites of the abuta plant (Cissampelos pareira Linn).
Target receptor analysis was carried out to see the receptor profile that will be used in this study by considering the main protease and RNA Dependent RNA Polymerase type of COVID-19 receptors; receptors having a resolution < 2Å; and receptors that have native ligand in the form of medicinal compounds on the PDB website accessed through https://www.rcsb.org and a good target protein analysis was performed using the Ramachandran plot.
The receptors are downloaded via the PDB website accessed at https://www.rcsb.org in “pdb” format. Then the receptor is optimized using YASARA to remove water molecules and to add hydrogen atoms. Then, the native ligand and protein were separated using YASARA. Proteins that have been separated from their ligands are stored in the “mol2” format [5].
The ligand was protonated at pH 7.4 and conformed to 10 conformers using MarvinSketch. The conformation results are stored in the form of “mol2” [5].
46 secondary metabolites in abuta (Cissampelos pareira Linn) were analyzed using a drug scan to determine which compounds could be proposed as drug candidates that could be administered orally based on Lipinski's rule of five [10]. Lipinski's rule of five parameters, including molecular weight < 500 g/mol, lipophilicity (log P) < 5, acceptor hydrogen bond < 10, and donor hydrogen bond < 5 [11]. The website used for drug scan analysis is SwissDock which can be accessed through http://www.swissadme.ch.
To show the capability of molecular docking simulations by re-tethering native ligands to receptors with the PLANTS application. The RMSD (Root Mean Square Deviation) is then calculated using YASARA software. The docking method is considered valid if the RMSD value is 2.0Å [12].
The docking process between ligands and proteins has been validated using the PLANTS application. The docking results appear in Excel form, indicating which conformation of the protein and ligand docking shows the lowest ligand-receptor binding docking score [5].
The analysis of the docking results was done by looking at the output in the form of the best rank shown in the Excel file from the docking results. The docking result is determined by selecting the ligand with the lowest bond docking score from the best score result.
Visualization of the docking was carried out to see amino acids or enzymes that make the stability of these compounds bound to their receptors [5]. The interaction was represented in two dimensions using the MOE.2009 program.
Prediction of ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) of secondary metabolites in abuta (Cissampelos pareira Linn) using the pkCSM website which can be accessed through https://biosig.lab.uq.edu.au/pkcsm/.
Molecular dynamics was carried out using the AMBER Google colab website. The protein used is the result of docking preparation which was re-prepared using YASARA and stored in the form of "pdb.". The ligands used include native ligands from each receptor, comparative drug compounds, and abuta (Cissampelos pareira Linn) secondary metabolite compounds that performed well on all receptors. The ligands were created with YASARA and stored as “pdb”.
The secondary metabolite of abuta (Cissampelos pareira Linn) was predicted for its synthesis pathway using the Chemical. AI website by considering several criteria, namely having a good docking score on all receptors, meeting Lipinski's rule of five criteria, and having a good ADMET predictive value.
The results of the analysis that met the requirements were 7AAP, 7D4F, 7BV2, 7CYQ, 7DFH, 7DOK, 5R7Y, 7JKV, 7TLL, and 7VH8. Receptors belonging to the main protease type are 5R7Y, 7JKV, 7TLL, and 7VH8. The receptors that belong to the type of RNA Dependent RNA Polymerase are 7AAP, 7D4F, 7BV2, 7CYQ, 7DFH, and 7DOK.
The receptors were then evaluated using a Ramachandran plot. A protein is considered a suitable target if the disallowed region of its non-glycine residue has a value less than 15% [13]. The ten receptors meet the criteria of having a non-glycine disallowed value of less than 15%, as shown in Table 1 and Figure 1.
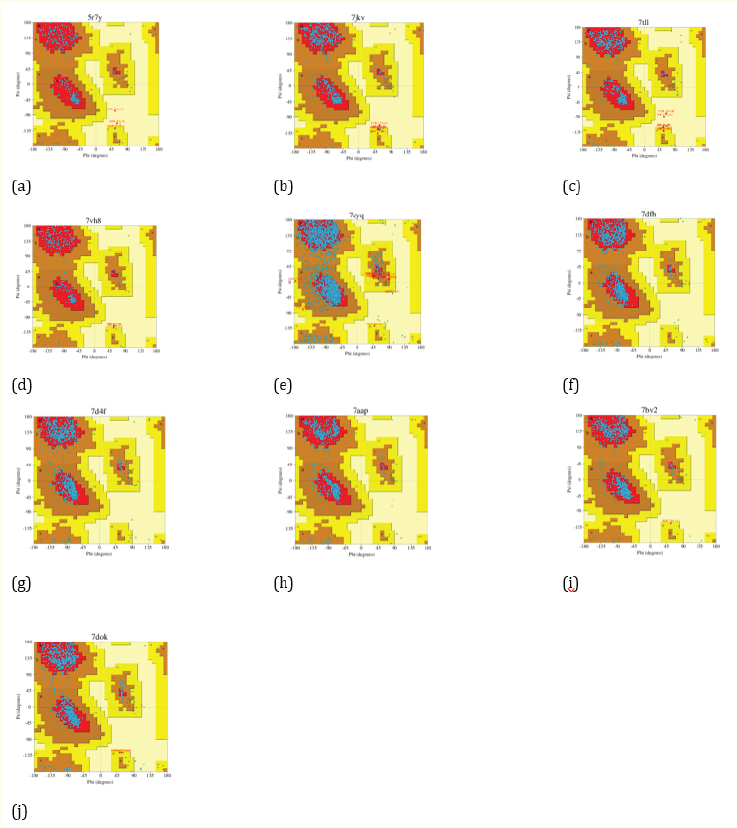
Figure 1: Ramachandran’s plot for receptors 5R7Y (a), 7JKV (b), 7TLL (c), 7VH8 (d), 7CYQ (e), 7DFH (f), 7D4F (g), 7AAP (h), 7BV2 (i), and 7DOK (j).
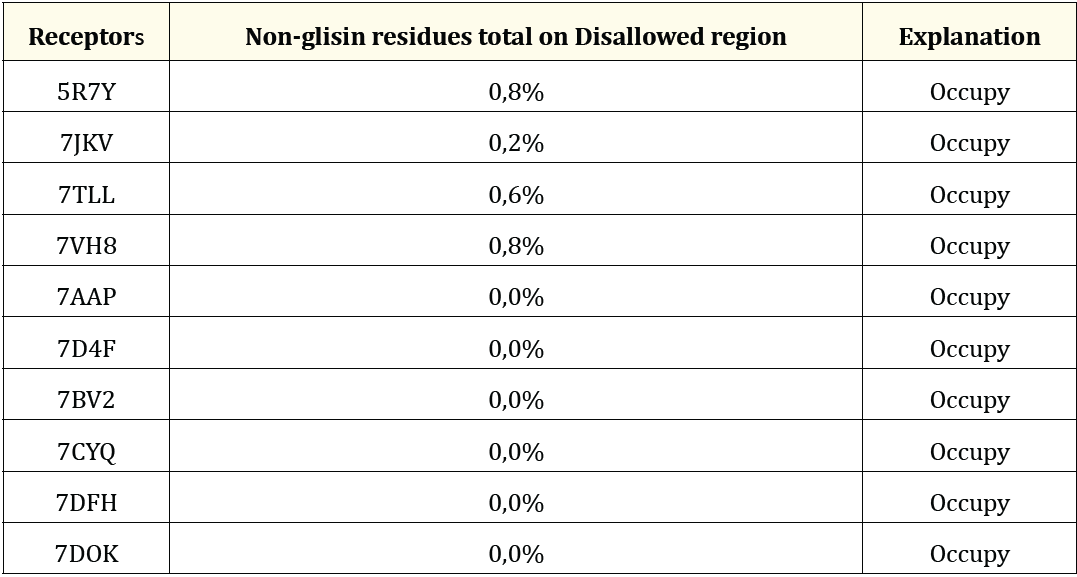
Table 1: Ramachandran’s plot analysis.
The 7AAP, 7D4F, 7BV2, 7CYQ, 7DFH, 7DOK, 5R7Y, 7JKV, 7TLL, and 7VH8 receptors were then downloaded from the PDB in “.pdb” format. The receptors are optimized by removing the interfering molecule, namely water and adding hydrogen to the receptor. Water needs to be removed from the receptor because the receptor downloaded from PDB still contains water, the water will interfere with the docking process and may interact with the compound [12]. The addition of hydrogen atoms into the system aims to facilitate crystal resolution in predicting the presence of hydrogen because the resolution of the crystal structure is not able to predict the presence of hydrogen in the system 5. Then, the receptor is stored in the form of a "yasara object". The file is opened with YASARA and then the native ligand and other interfering molecules are removed so that only proteins are left. The protein is stored in the “mol2” format.
After the receptor is prepared and stored in the form of a “yasara object”, the file “yasara object” is reopened using the YASARA. Then the unused molecules including the protein are removed so that only the native ligand remain. Then the native ligand are stored in the “mol2” form. After that, this native ligand was protonated at pH 7.4 and made into 10 conformers using MarvinSketch [5].
A total of 46 secondary metabolites of abuta (Cissampelos pareira Linn) and 4 comparison drug compounds were prepared using MarvinSketch by copying the SMILES of these compounds from the PubChem website, then pasting them on MarvinSketch. Then the compound is protonated at pH 7.4 to condition the body's pH [14]. The purpose of protonation also is to add atomic charge to the compound and can see the hydrogen bonds that occur [15]. Then, the ligands were conformed to 10 conformers to determine in which conformation the compound produced the lowest docking score [14].
There are 42 secondary metabolites of abuta plant (Cissampelos pareira Linn) that meet Lipinski's rule of five which indicates that these compounds can be proposed as candidates for drugs given orally. These compounds are Pareirarine, Nor-N-Magnoflorine, Cissamine, Magnocurarine, Oblongine, Coclaurine, Nuciferine, Corytuberine, Quercitol, Thymol, Laudanosine, Berberine, Dicentrine, Hayatine, Pelosine, Hayatinine, Isochondodendrine, Cissarandine, Insularine, Cycleante, Sepeerine, Obaberine, Obamegine, Homoaromoline, Nor-N-Chondrocurine, 1-(4-(formyloxy)-3-methoxybenzyl)-6,7-dimethoxy-2,2-dimethyl1,2,3,4-tetra-hydro-isoquinolin2-ium, Dehydrodicentrine, Cissampeline, Trans-N-Feruloyltyramine, Salutaridine, Bulbucarpine, Cissampeloflavone, Hayatidine, Pareirubrine A, Pareirubrine B, Isoimerubrine, Grandirubrine, Parei-tropone, Norimeluteine, Norruffscine, and Magnoflorscine. Then, there are 4 secondary metabolite compounds that do not meet Lipinski's rule of five which indicates that these compounds cannot be proposed as drug candidates given orally, but can be given through another route, namely parenteral. These compounds are Amentoflavone, Reserpine, Kaempferol-3-O-β-D-Glucopyranoside, and Kaempferol-3-O-β-D-Glucuronopyranoside.
The condition for receiving receptor validation is the docking result RMSD < 2Å [12]. To see the RMSD redocking results between native ligand and receptors, use YASARA by entering the results of the ligands that have the best docking scores based on the results of docking native ligand redocked with native ligand that have not been redocked into YASARA, then remove the hydrogen in these two molecules and stored in the form of “.sce”. In YASARA it is not allowed to shift the molecule because it can change the 3D coordinates of the molecule so that the RMSD value will be invalid [5]. As a result, only 4 receptors had RMSD values < 2Å. These receptors belong to the main protease receptor type and the receptors are 5R7Y with an RMSD value of 1.7493Å; 7JKV with an RMSD value of 0.7154Å; 7TLL with RMSD value of 1.0349Å; and 7VH8 with an RMSD value of 1.0671Å. None of the RdRp receptors showed RMSD values < 2Å, so only the main protease receptor was used in this study. The results of the 5R7Y, 7JKV, 7TLL, and 7VH8 receptor validation are shown in Figure 2.
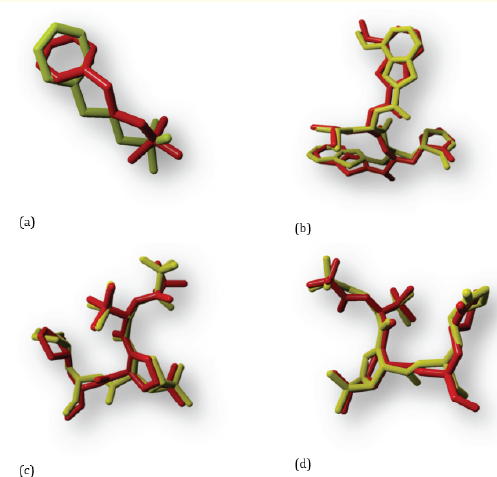
Figure 2: Results of native ligand (yellow) overlaps with the unredocking native ligand (red) at the 5R7Y (a), 7JKV (b), 7TLL (c), and 7VH8 (d) receptors.
The redocking results between the native ligand and the 5R7Y receptor showed a docking score of -64.76. The results of docking the secondary metabolite compound of abuta plant (Cissampelos pareira Linn) to the 5R7Y receptor showed that the compound that had a lower docking score than the native ligand was 29 compounds. The compound that has the lowest docking score is Isochondodendrine compound with a docking score of -94.60 and the compound that has the highest docking score is Laudanosine compound which is -65.12. Then, the score of Favipiravir docking with the 5R7Y receptor was -53.15. Compounds that have a lower docking score than Favipiravir are 39 compounds. The compound that had the lowest docking score was Isochondo-dendrine with a score of -94.60; while the compound that had the highest docking score was Pareirubrine A compound with a score of -53.19.
The docking result between Molnupiravir and the 5R7Y receptor is -77.56. Compounds that had a lower docking score compared to Molnupiravir were 7 compounds. The compound that had the lowest docking score was Isochondodendrine with a docking score of -94.60; while the compound that has the highest docking score is Amentoflavone compound with a score of -80.05. However, neither compound had a lower docking score than Nirmatrelvir and Remdesivir. This is because the score of Nirmatrelvir docked with the 5R7Y receptor is -95.08 and the score of Remdesivir docked with the 5R7Y receptor is -107.42. The results of docking secondary metabolites of abuta plant (Cissampelos pareira Linn) on the 5R7Y receptor did not have a lower score than the two comparison drug compounds.
The redocking result between the native ligand and the 7JKV receptor was -116.5. Meanwhile, the docking results for abuta secondary metabolites (Cissampelos pareira Linn) at the 7JKV receptor showed that none of the compounds had a lower docking score than the native ligand where the compound with the lowest docking score was Cissampeline with a docking score of -94.87; the value is not lower than -116.5. Then the result of docking Favipiravir with the 7JKV receptor was -59.37 which indicated that there were 43 compounds that had a lower docking score than Favipiravir. The compound that had the lowest docking score was Cissampeline with a score of -94.87 and the compound that had the highest docking score was Nuciferine with a score of -60.64.
The docking result of Molnupiravir with the 7JKV receptor was -83.91. Secondary metabolite compounds that have a lower docking score than Molnupiravir are 10 compounds. The compound that had the lowest docking score was Cissampline with a score of -94.87, and the compound that had the highest docking score was Coclaurine with a score of -86.31. The docking result of Nirmatrelvir with the 7JKV receptor was -96.09 and the docking result of Remdesivir with the 7JKV receptor was -120.61 and there was no abuta secondary metabolite compound (Cissampelos pareira Linn) which had a lower docking score than the two comparison drug compounds, so that the secondary metabolite abuta (Cissampelos pareira Linn) could not exceed the docking score of the two comparison drug compounds at the 7JKV receptor.
The result of redocking between native ligand and 7TLL receptor is -96,17. There are 2 compounds that have lower docking scores compared to natural ligands. The compound that had the lowest docking score was Kaempferol-3-O-β-D-Gluco-pyranoside with a docking score of -99.03 and the compound that had the highest docking score was Cissampeline with a docking score of -97.20. Then for the docking result between Favipiravir and the 7TLL receptor is -56.39 so that there are 44 compounds that have a lower docking score than Favipiravir. The compound that had the lowest docking score was Kaempferol-3-O-β-D-Glucopyranoside with a score -99.03 and the compound that had the highest docking score was Norimeluteine with a docking score of -58.74.
The docking score between Molnupiravir and the 7TLL receptor was -84.21 and there were 13 compounds that had a lower docking score than Molnupiravir. The compound that had the lowest docking score was Kaempferol-3-O-β-D-Glucopyranoside with a docking score of -99.03 and the compound that had the highest docking score was Corytuberine with a score of -84.30. The docking result between Nirmatrelvir and the 7TLL receptor was -105.85 and the docking result between Remdesivir and the 7TLL receptor was -120.37. The lowest docking score resulting from the docking of the secondary metabolite compound of abuta (Cissampelos pareira Linn) was -99.03, and none of the secondary metabolites of abuta (Cissampelos pareira Linn) had a lower docking score than the two comparison drug compounds, so that the metabolite compound Abuta secondary (Cissampelos pareira Linn) could not exceed the docking scores of the two comparison drug compounds at the 7TLL receptor.
The redocking result between the native ligand and the 7VH8 receptor was -102.23, while the lowest score for the secondary metabolite compound Abuta (Cissampelos pareira Linn) was -97.72 (Cissampeline). So that there is no abuta secondary metabolite compound that has a lower docking score than the natural 7VH8 ligand. The result of docking Favipiravir with 7VH8 receptors is -55.19 so there are 43 secondary metabolites of abuta (Cissampelos pareira Linn) which have a lower docking score than Favipiravir. The compound that had the lowest docking score was Cissampline with a docking score of -97.72 and the compound that had the highest docking score was Isoimerubrine with a score of -58.16. Then, the docking result of Molnupiravir with the 7VH8 receptor was -82.54 so there were 11 compounds that had a lower docking score than Molnupiravir. The compound that had the lowest docking score was Cissampeline with a score of -97.72 and the compound that had the highest docking score was Corytuberine with a score of -83.48. The docking score between Nirmatrelvir and the 7VH8 receptor was -107.47, and the docking score of Remdesivir with the 7VH8 receptor was -118.12 and none of the secondary metabolites abuta (Cissampelos pareira Linn) had a lower docking score than the two comparison drugs. Thus, the secondary metabolite compound abuta (Cissampelos pareira Linn) could not exceed the docking score of the two comparison drug compounds at the 7VH8 receptor.
The visualization of the docking results aims to show the bonds formed between the compound (ligand) and the amino acids found in the protein (receptor), which contribute to the substance’s stability of the compound [5]. This visualization was also carried out to determine the active site of the receptor in binding compounds [16]. Hydrogen bonds are bonds that play a crucial role in the interaction between the ligand and the receptor, contributing to the ligand’s affinity for the target receptor [17].
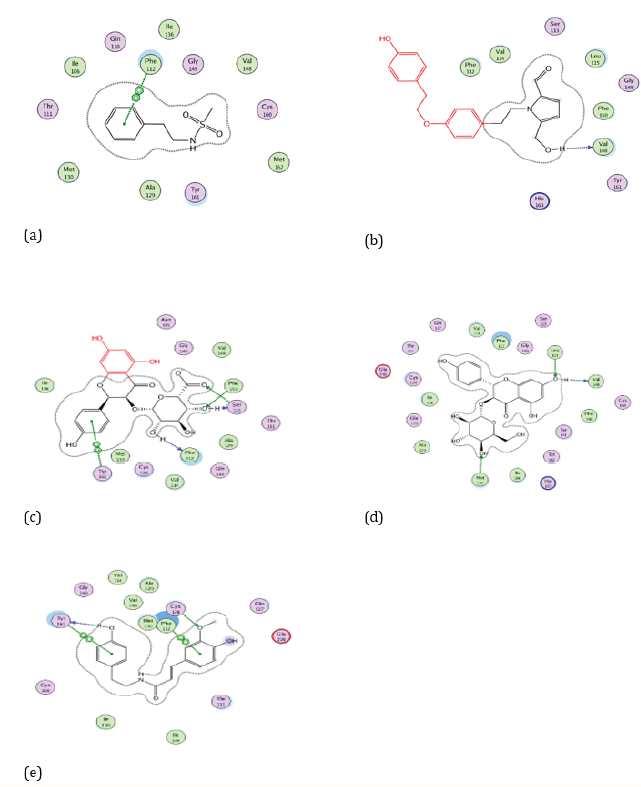
Figure 3: Docking visualization of native ligand (a), Cissampeline (b), Kaempferol-3-O-β-D-Glucopyranoside (c), Kaempferol-3-O-β-DGlucuronopyranoside (d), and Trans-N-Feruloyltyramine (e) with 5R7Y.
Based on Figure 3. the native ligand of the 5R7Y receptor has a hydrogen bond on the amino acid residue of Phe112. While the Cissampline compound has hydrogen bonds at the amino acid residue of the 5R7Y receptor, namely at Val148. Then the compound Kaempferol-3-O-β-D-Glucu-ronopyranoside has hydrogen bonds at the 5R7Y amino acid residue, namely Phe112 and Phe150. The compound Kaempferol-3-O-β-D-Glucopyranoside has hydrogen bonds at the 5R7Y amino acid residue, namely Leu115, Val148, and Met130. The Trans-N-Feruloyltyramine compound has hydrogen bonds at the amino acid residue of the 5R7Y receptor, namely Phe112. Compounds that have the same hydrogen bonding sites as natural ligands, namely the amino acid residue of Phe112 are Kaempferol-3-O-β-D-Glucuronopyrano-side and Trans-N-Feruloyltyramine compounds.
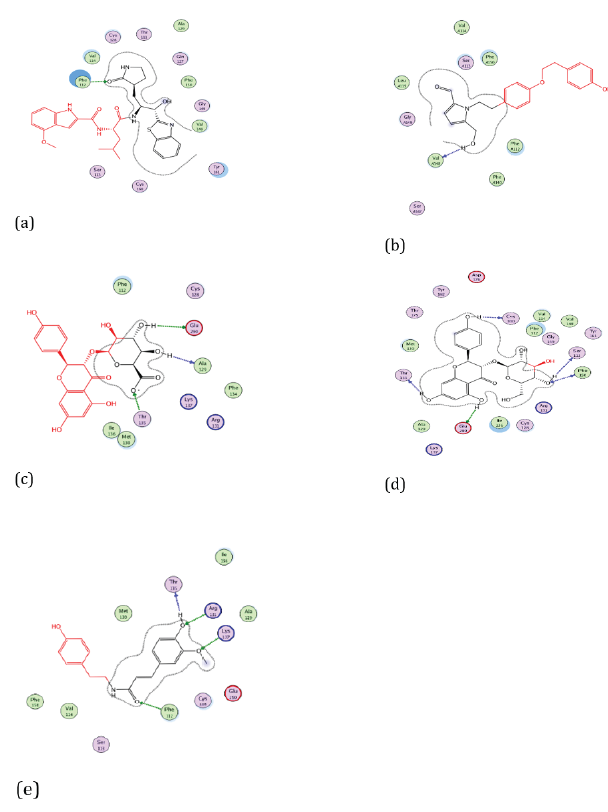
Figure 4: Docking visualization of native ligand (a), Cissampeline (b), Kaempferol-3-O-β-D-Glucopyranoside (c), Kaempferol-3-O-β-DGlucuronopyranoside (d), and Trans-N-Feruloyltyramine (e) with 7JKV.
Based on Figure 4. the native ligand of the 7JKV receptor has hydrogen bonds at the amino acid residues Phe112, and Val148. Meanwhile, Cissampline has hydrogen bonds at the amino acid residue of the 7JKV receptor, namely Val148. Then the compound Kaempferol-3-O-β-D-Glucuronopyranoside has a hydrogen bond at the amino acid residue of the 7JKV receptor, namely Ala129. The compound Kaempferol-3-O-β-D-Glucopyranoside has hydrogen bonds at the amino acid residues of Phe150. The Trans-N-Feruloyltyramine compound has hydrogen bonds at the amino acid residue of the 7JKV receptor, namely Phe112. Compounds that have the same hydrogen binding site as the native ligand 7JKV, namely in Val148 are Cissampeline compounds. Meanwhile, the compound which has the same hydrogen binding site as the natural ligand, namely Phe112 is the Trans-N-Feruloyltyramine compound.
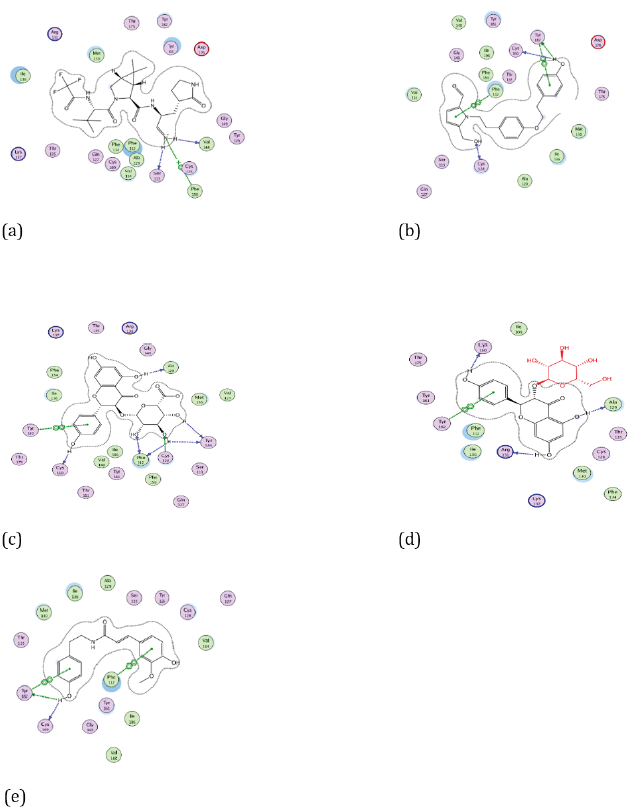
Figure 5: Docking visualization of native ligand (a), Cissampeline (b), Kaempferol-3-O-β-D-Glucopyranoside (c), Kaempferol-3-O-β-DGlucuronopyranoside (d), and Trans-N-Feruloyltyramine (e) with 7TLL.
Based on Figure 5 the native ligand of the 7TLL receptor has hydrogen bonds to the amino acid residues Val148, and Phe150. While the Cissampline compound has hydrogen bonds at the amino acid residue of the 7TLL receptor, namely Phe112. Then the compound Kaempferol-3-O-β-D-Glucuronopyranoside has hydrogen bonds at the amino acid residues of the 7TLL receptor, namely Phe112, and Ala129. The compound Kaempferol-3-O-β-D-Glucopyranoside has a hydrogen bond at the amino acid residue 7TLL, namely Ala129. The Trans-N-Feruloyltyramine compound has hydrogen bonds at the amino acid residue of the 7TLL receptor, namely Phe112.
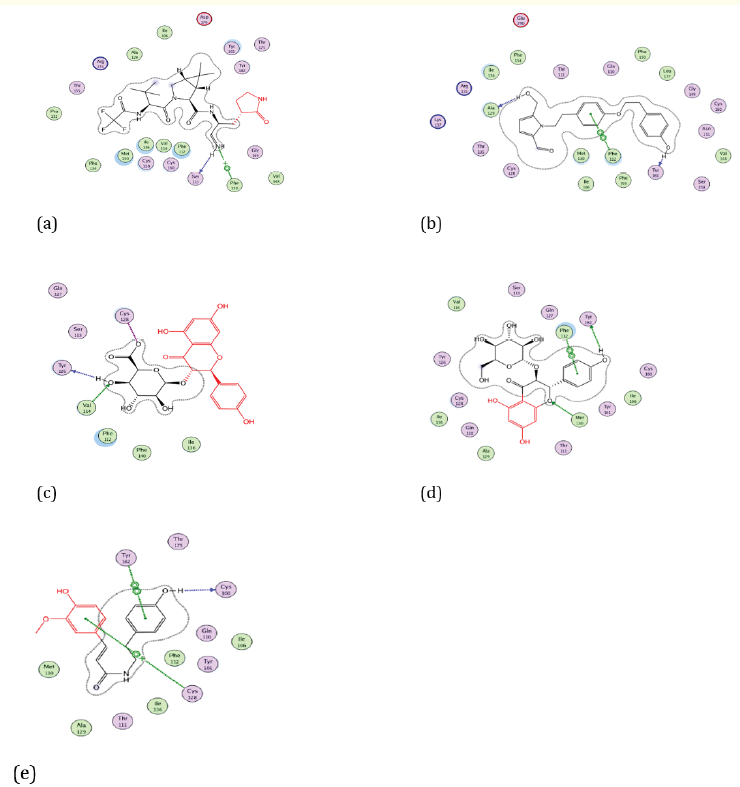
Figure 6: Docking visualization of native ligand (a), Cissampeline (b), Kaempferol-3-O-β-D-Glucopyranoside (c), Kaempferol-3-O-β-DGlucuronopyranoside (d), and Trans-N-Feruloyltyramine (e) with 7VH8.
Based on Figure 6 the native ligand of the 7VH8 receptor has a hydrogen bond on the amino acid residue at Phe150. While the secondary metabolite compound abuta (Cissampelos pareira Linn) namely Cissampeline has hydrogen bonds with the amino acid residues of Phe112 and Ala129. Then the compound Kaempferol-3-O-β-D-Glucuronopyranoside has a hydrogen bond on the amino acid residue Val114. The compound Kaempferol-3-O-β-D-Gluco-pyranoside has conventional hydrogen bonds at the amino acid residue 7VH8, namely Phe112, and Met130. The Trans-N-Feruloyltyramine compound does not have a hydrogen bond at the amino acid residue of the 7VH8 receptor.
Based on the description above, three secondary metabolites that have good docking scores at the four receptors (5R7Y, 7JKV, 7TLL, 7VH8) are Cissampeline, Kaempferol -3-O-β-D-Glucuronopyrano-side, Kaempferol-3-O-β- D-Glucopyrano-side, and Trans-N-Feruloyltyramine. These compounds were supported by ADMET prediction data which were analyzed using the pkCSM website which can be seen in Table 3 Prediction of ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) was performed using the pkCSM application. The models used to predict absorption are Caco-2 permeability, water solubility, and intestinal absorption. Caco-2 is a cell layer consisting of adeno-carcinoma colorectal epithelial cells. Caco-2 is widely used to model in vitro studies of human intestinal mucosa to predict absorption of orally administered drugs. Compounds that have a predicted Caco-2 value > 0.90 are said to have a high Caco-2 absorption value. Drugs taken orally will be absorbed in the small intestine, drug molecules are said to be low absorption if the value is less than 30%. For the water solubility model, which shows the value of the drug dissolved in water at a temperature of 25o C, the value is expressed in units of log mol/L. The models used to predict drug distribution are VDss and CNS permeability. VDss is the volume of distribution of the drug at a theoretically distributed total dose giving the same concentration in the body. The VDss value is said to be low if < -0.15 log L/kg, and high if the value is > 0.45 log L/kg. CNS (Central Nervous System) permeability or permeability in the CNS (Central Nervous System) is a model to determine the distribution of drugs in the brain directly or more easily. A compound is said to be well concentrated in the brain if the logPS (log permeability surface) value is more than -2, and a compound is said to be unable to penetrate the CNS well if its logPS value is less than -3.
Prediction of the metabolism of a drug compound in pkCSM using the cytochrome P-450 model. A drug compound will be metabolized by cytochrome P-450 enzymes which are mostly found in the liver which is a xenobiotic oxidizing enzyme for the purpose of excretion of a compound. Many compounds can be activated by this enzyme and some compounds can also be activated by this enzyme. Cytochrome P-450 inhibitors such as grapefruit juice may affect drug metabolism and are contraindicated. For this reason, it is important to know what compounds can inhibit this cytochrome P-450 enzyme. The models used in pkCSM for the different isoforms were CYP1A2/CYP2CI9, CYP2C9/CYP2D6/CYP3A4. A compound is said to be a cytochrome P-450 inhibitor if the concentration required to cause inhibition is less than 50%. Prediction of excretion using the total clearance model. Total clearance is a combination of renal clearance and hepatic clearance expressed in units of log(ml/min/kg). Prediction of compound toxicity using a hepatotoxicity model and LD50 whether the compound can cause hepatotoxicity and what the LD50 value is [18]. Because the D50 value in pkCSM is expressed in moles/kg, it needs to be converted to mg/kg to make it easier to identify the toxicity category. The toxicity categories are shown in Table 2.
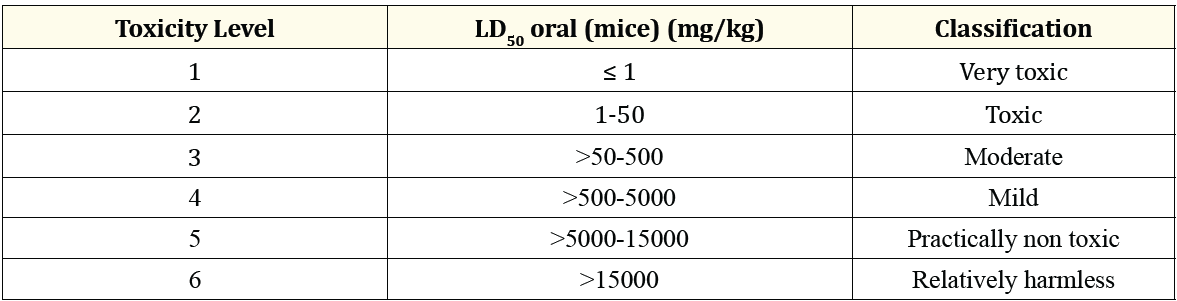
Table 2: Classification of the toxicity test preparations. (Source: Siswandi and Saragih, 2018).
Table 3 the results of the predicted absorption of Cissampeline compounds can be absorbed in the intestine by 93.56%, which means that Cissampeline compounds can be well absorbed in the small intestine. Cissampeline compound has a water solubility of -4.6 log mol/L; and the permeability of Caco-2 is 1.012 log cm/s which indicates that the compound has a high permeability value. Meanwhile, based on the predicted distribution, Cissampeline compound has a VDss value of 0.254 log L/kg which indicates the compound has a good distribution value in the blood; and the CNS permeability value of -2,654 which indicates that Cissampeline can penetrate well in the CNS/CNS. Cissampelin compounds are metabolized by CYP3A4 substrate enzymes, CYP2C19 inhibitors, CYP1A2 inhibitors, and CYP2C9 inhibitors. Then, Cissampaline compound has a total clearance value of 0.48 log(ml/min/kg). And the Cissampline compound is not hepatotoxic, and has an LD50 value of 1,984 mol/kg which when converted into mg/kg becomes 724,160 mg and is in the relatively harmless range based on Table 2.
Table 3 the predicted absorption of the compound Kaempferol-3-O-β-D-Glucuronopyranoside has a water solubility of -2.62 log mol/L; the permeability value of Caco-2 is -0.74 log cm/s which indicates that the compound Kaempferol-3-O-β-D-Glucuronopyrano-side has poor permeability in Caco-2 cells; and compounds Kaempferol-3-O-β-D-Glucuronopyranoside can be absorbed as much as 13.2% which indicates that these compounds can be poorly absorbed in the small intestine. Then the compound Kaempferol-3-O-β-D-Glucuronopyrano-side has a VDss value of 0.66 log L/kg which indicates that the compound can be moderately distributed in the blood; and has a CNS/CNS permeability value of -4.05 which indicates that the compound cannot penetrate well in the CNS/CNS. The compound Kaempferol-3-O-β-D-Glucu-ronopyranoside does not inhibit metabolic enzymes in the liver. Then, the compound Kaempferol-3-O-β-D-Glucuronopyrano-side has a total clearance value of -0.17 log(ml/min/kg). The compound Kaempferol-3-O-β-D-Glucuronopyrano-side does not cause hepatotoxicity, and has an LD50 value of 2.49 mol/kg which when converted into mg/kg is 1.156.306 mg/ kg which if based on Table 2. the value is included in the category of relatively harmless.
Table 3 the absorption prediction of Kaempferol-3-O-β-D-Glucopyranoside compounds was absorbed as much as 35.88% in the intestine which indicates that this compound is poorly absorbed in the small intestine; has a solubility in water of -3.45 log mol/L; and the permeability in Caco-2 was 0.55 log cm/s which indicated that the Kaempferol-3-O-β-D-Glucopyranoside compound had poor permeability in Caco-2 cells. The compound Kaempferol-3-O-β-D-Gluco-pyranoside has a VDss value of 0.02 log L/kg which indicates that the compound is well distributed in the blood. Then the compound Kaempferol-3-O-β-D-Gluco-pyranoside has a CNS/CNS permeability value of -4.61 which indicates that the compound cannot be penetrated in the CNS/CNS.
The compound Kaempferol-3-O-β-D-Glucopyranoside did not inhibit metabolic enzymes in the liver, and had a total clearance value of 0.2 log(ml/min/kg). The compound Kaempferol-3-O-β-D-Gluco-pyranoside does not cause hepatotoxicity, and has an LD50 value of 2.71 mol/kg which when converted into mg/kg is 1,220,556 mg/kg and based on Table 2. the value is included in the category of relatively harmless.
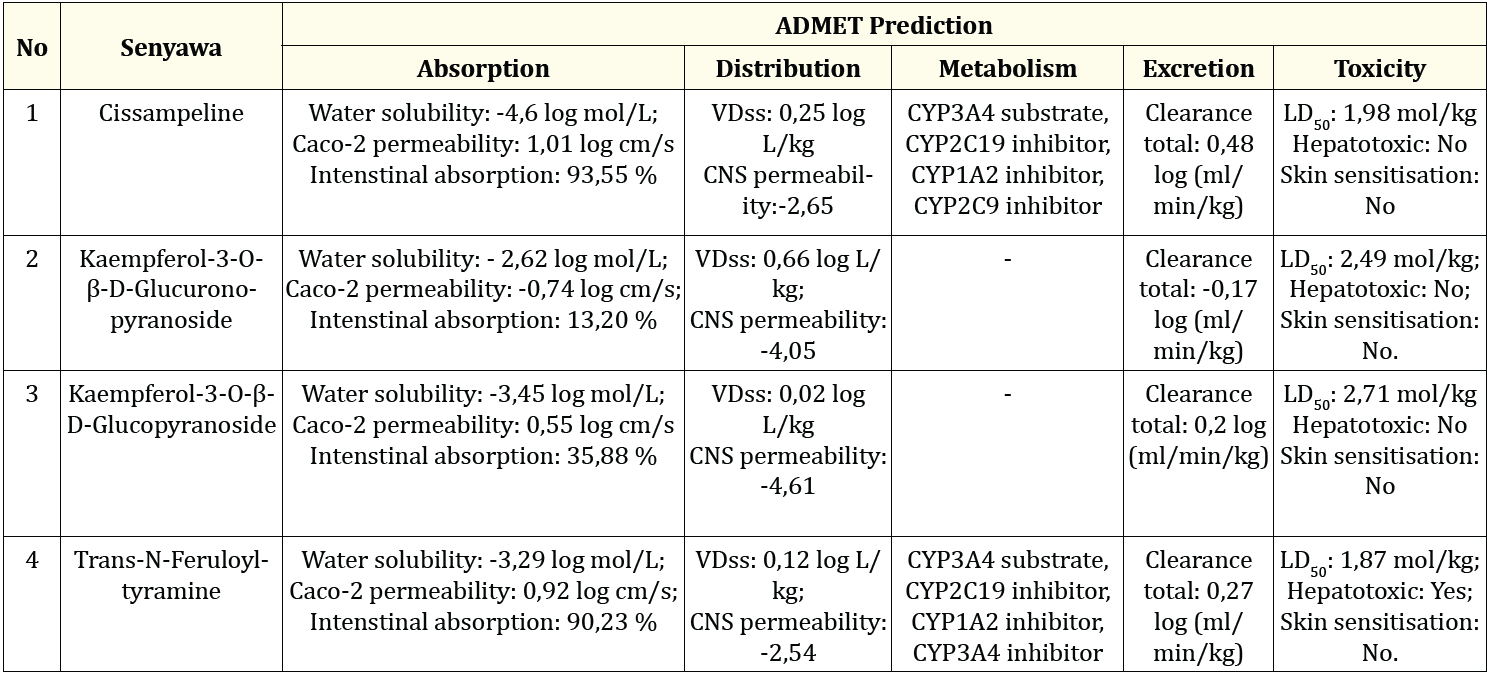
Table 3: Presents the predicted of ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) analysis.
Trans-N-Feruloyltyramine compounds are metabolized by CYP3A4 substrate enzymes, CYP2C19 inhibitors, CYP1A2 inhibitors, and CYP3A4 inhibitors. Then, the compound Trans-N-Feruloyltyramine has a total clearance value of 0.27 log(ml/min/kg). However, the Trans-N-Feruloyltyramine compound is hepatotoxic, and has an LD50 value of 1.87 mol/kg which when converted into mg/kg form becomes 586,904 mg and is in the relatively harmless range based on in Table 2. The compound Trans-N-Feruloyltyramine did not cause skin sensitisation.
Secondary metabolite compound of abuta (Cissampelos pareira Linn) which has a good docking score at 4 receptors (5R7Y, 7JKV, 7TLL, and 7VH8); comply with Lipinski's rule of five; and has a good predictive value of ADMET by molecular dynamics analysis and the compound is Cissampeline. The peptide structure in molecular dynamics is not rigid so that it can move according to its flexibility and its movement is not limited to rotation so that the molecular dynamics modeling can provide information on structural, dynamics, and energy stability [20].
Molecular dynamics simulation was carried out at a temperature of 312 K where the temperature when converted into Celsius units is 38oC. This temperature is the temperature of the human body when you have a fever. One of the symptoms of COVID-19 is fever [21] so the temperature was chosen to condition the system according to the temperature of the human body when experiencing a fever. The comparison drug compound used to compare this molecular dynamics simula-tion is Molnupiravir. The simulation stage includes 1000 steps of energy minimization, heating to a temperature of 312 K, temperature equilibration, pressure equilibration, and the simulation process starts for 10 ns (nanosecond). The results of the molecular dynamics analysis are the RMSD (Root Main Square Deviation), RMSF (Root Main Square Fluctuation) values, and the interaction between the protein and the ligand.
The stability and rationality of compound-receptor interactions can be determined by examining the RMSD values. The ligand binding to the receptor is considered steady and eligible if the average RMSD value is less than 2Å [22]. Based on Figure 7. the average RMSD of Cissampline compounds is 1.77Å; while the average RMSD of native ligand is 1.33Å; and the mean RMSD of Molnupiravir was 1.62Å. So that the three compounds are said to be stable in binding to the 5R7Y receptor at a simulation time of 10 nanoseconds.
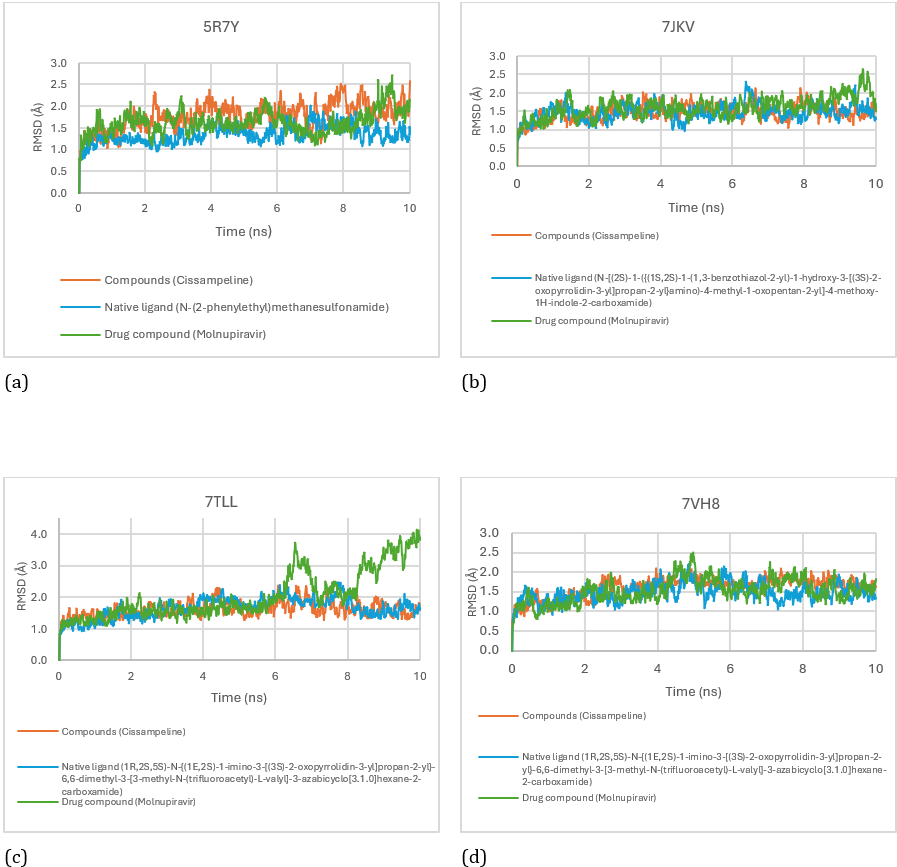
Figure 7: RMSD Score for Cissampeline, Native Ligand, and Molnupiravir on receptors 5R7Y (a), 7JKV (b), 7TLL (c), and 7VH8 (d).
Based on Figure 7. the average RMSD of Cissampline compounds at the 7JKV receptor is 1.45Å; while the average RMSD of native ligand is 1.46Å; and the mean RMSD of Molnupiravir was 1.59Å. So that the three compounds are said to be stable binding to the 7JKV receptor at a simulation time of 10 nanoseconds. The average RMSD of Cissampeline compounds at the 7TLL receptor was 1.59Å; while the average RMSD of native ligand is 1.65Å; and the mean RMSD of Molnupiravir was 2.02Å. So that the native ligand and Cissampeline compounds are said to be stable to bind to the 7TLL receptor at a simulation time of 10 nanoseconds. Meanwhile, the comparison drug compound, Molnupiravir, was said to be unstable in molecular dynamics simulations for 10 nanoseconds because the RSMD average value was > 2Å. The binding between molnupiravir and the 7TLL receptor was unstable starting at a simulation time of 5.68 nanoseconds with an RMSD value of 2.09Å. While the average RMSD of Cissampeline compounds at the 7VH8 receptor was 1.62Å; the average RMSD of native ligand is 1.46Å; and the mean RMSD of Molnupiravir was 1.53Å. So that the three compounds are said to be stable in binding to the 7VH8 receptor at a simulation time of 10 nanoseconds.
To find out fluctuations in the interaction between the ligand and the receptor during the simulation, it can be seen through the RMSF results. RMSF measures the amount of variability in the movement of each residue during the simulation; the lower the RMSF number, the more stable the connection between the ligand and its receptor [23]. Based on Figure 8. there is fluctuation in the interaction between the ligand and the 5R7Y receptor. The Cissampeline compound binding area at the 5R7Y receptor as the lowest pocket was Leu:27, Val:204, and Asn:203 residues which indicated that in that area the Cissampline compound and 5R7Y receptor binding were stable with an average RMSF value of 0.88Å. The residues that bind to native ligand in the lowest pocket are Leu:27, Val:204, and Asn:203 which indicate that in these areas the native ligand binding to the 5R7Y receptor is stable with an average RMSF value of 0.88. The residues that bind to Molnupiravir in the lowest pocket are Cys:38, Val:204, and Asn:203 which indicate that in these areas the binding of Molnupiravir to the 5R7Y receptor is stable with an average RMSF value of 0.99Å. The Cissampline compound has the same stable binding as the native ligand of the 5R7Y receptor.
Based on Figure 8. there is fluctuation in the interaction between the ligand and the 7JKV receptor. The area of the Cissampline compound binding to the 7JKV receptor as the lowest pocket was Leu:27, Val:204, and Asn:203 residues which indicated that the Cissampaline compound binding area and the 7JKV receptor were stable with an average RMSF of 0.91Å. The residues that bind to native ligand in the lowest pocket are Cys:38, His:38, and Val:204 which indicate that in these areas the native ligand binding to the 7JKV receptor is stable with an average RMSF value of 0.84Å. The residues binding to Molnupiravir in the lowest pocket were Leu:27, Cys:38, and Val:204, which indicated that in these areas the binding of Molnupiravir to the 7JKV receptor was stable with an average RMSF value of 0.96Å. Then there is a fluctuation in the interaction between the ligand and the 7TLL receptor. The region of Cissampaline compound binding to the 7TLL receptor as the lowest pocket was residues Gly:29, Tyr:37, and Val:36 which indicated that in that area the Cissampaline compound binding to the 7TLL receptor was stable with an average RMSF of 0.93Å. The residues that bind to native ligand in the lowest pocket are Leu:27, Tyr:37, and Val:204 which indicate that in these areas the native ligand binding to the 7TLL receptor is stable with an average RMSF of 0.98Å. The residues binding to Molnupiravir in the lowest pocket were Gly:29, Val:204, and Leu:205 which indicated that in these areas the binding of Molnupiravir to the 7TLL receptor was stable with an average RMSF value of 1.32Å. Then, there was fluctuation in the interaction between the ligand and the 7VH8 receptor. The Cissampaline compound binding area at the 7VH8 receptor as the lowest pocket was Cys:145, Ser:147; Tyr:37 which shows that in that area the Cissampaline compound binding to the 7VH8 receptor is stable with an average RMSF value of 0.95Å. The residues that bind to native ligand in the lowest pocket are Leu:27, Asn:203, and Val:204 which indicate that in these areas the native ligand binding to the 7VH8 receptor is stable with an average RMSF of 0.85Å. The residues that bind to Molnupiravir in the lowest pocket are Val:36, Tyr:37, and Val:204 which indicate that in these areas the binding of Molnupiravir to the 7VH8 receptor is stable with an average RMSF value of 1Å.
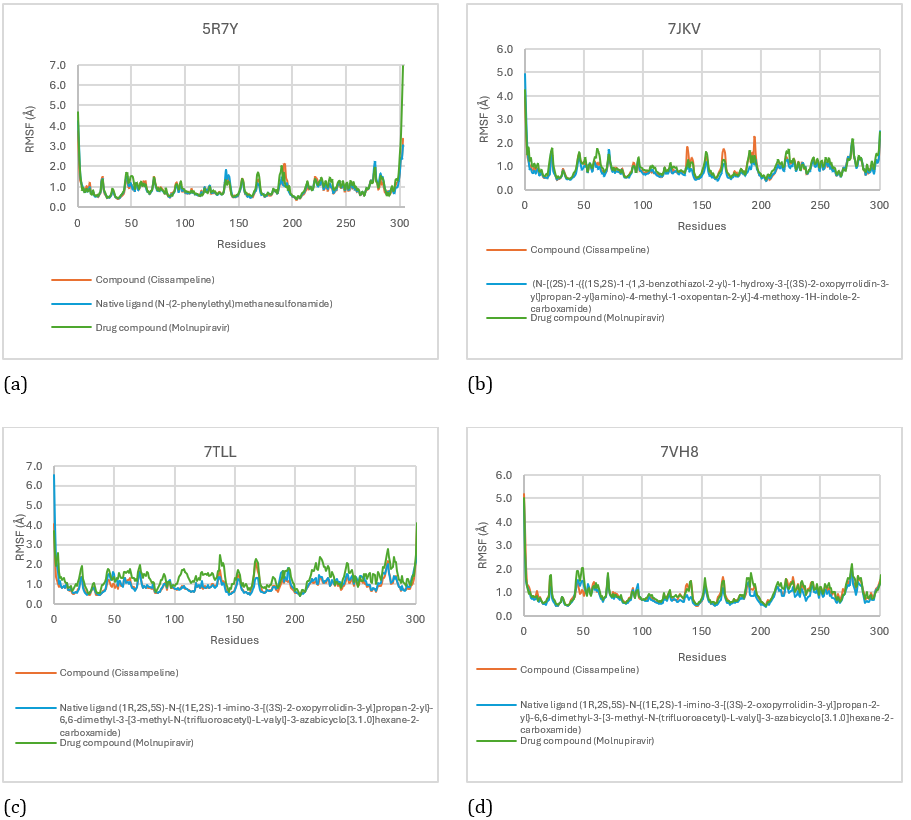
Figure 8: RMSF score for Cissampeline, Native Ligand, and Molnupiravir on receptors 5R7Y (a), 7JKV (b), 7TLL (c), and 7VH8 (d).
Molecular dynamics simulations at 312 K show multiple hydrogen bonds at the binding sites on each receptor. The stability of the ligand bond with the receptor is influenced by hydrogen bonding, the more residues that bind to the ligand, the stronger the ligand bond with the receptor [24].
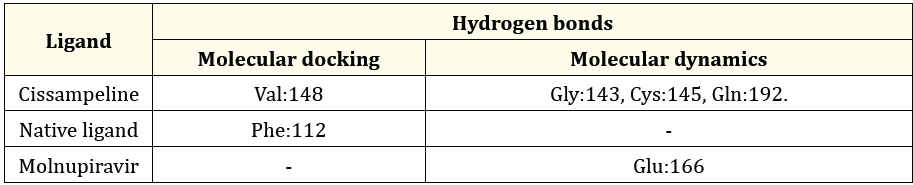
Table 4: The Comparison between hydrogen bonds formed during molecular docking and after molecular dynamics simulation on 5R7Y.
Table 4 the results of molecular docking of Cissampeline compounds have hydrogen bonds at the amino acid residue of the 5R7Y receptor, namely at Val:148. Meanwhile, the results of molecular dynamics simulations show that Cissampeline compounds have hydrogen bonds at the amino acid residues of the 5R7Y receptor, namely Gly:143, Cys:145, and Gln:192. The results of molecular docking of native ligand with 5R7Y receptors showed hydrogen bonds at the Phe:112 residue, whereas after molecular dynamics simulations showed that native ligand did not have hydrogen bonds. Then, based on the results of molecular docking between Molnupiravir and the 5R7Y receptor, no hydrogen bonds were shown. Meanwhile, based on the results of molecular dynamics simulations, it shows that Molnupiravir has hydrogen bonds with the 5R7Y receptor at the Glu:166 residue. Cissampeline compounds have more hydrogen bonds than native ligand with Molnupiravir so it can be said that Cissampeline compounds are more stable than native ligand and Molnupiravir.

Table 5: The Comparison between hydrogen bonds formed during molecular docking and after molecular dynamics simulation on 7JKV.
Based on Table 5 the results of molecular docking of Cissampeline compounds have hydrogen bonds at the amino acid residues of the 7JKV receptor, namely at Val:148. Meanwhile, the results of the molecular dynamics simulation showed that the Cissampeline compound had hydrogen bonds at the amino acid residue of the 7JKV receptor, namely Glu:166. The results of molecular docking of native ligand with the 7JKV receptor showed hydrogen bonds at Phe:112, and Val:148 residues, whereas after molecular dynamics simulations showed that native ligand had hydrogen bonds at Gln:189, Gly:143, Hie:163, and Hie: 164, Glu:166, and Phe:140. Then, based on the results of molecular docking between Molnupiravir and the 7JKV receptor, no hydrogen bonds were shown. Meanwhile, based on the results of molecular dynamics simulations, it shows that Molnupiravir has hydrogen bonds with the 7JKV receptor at the Glu:166 residue. Cissampeline compounds, natural ligands, and Molnupiravir have hydrogen bonds at the amino acid residue of the same 7JKV receptor, namely Glu:166.
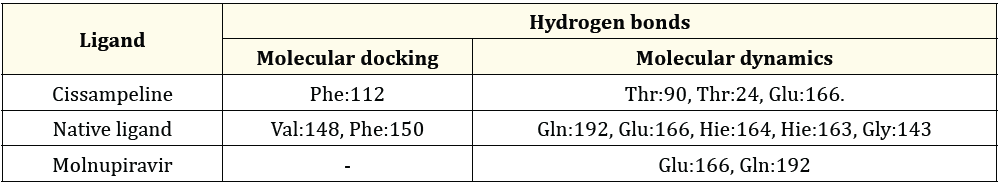
Table 6: The Comparison between hydrogen bonds formed during molecular docking and after molecular dynamics simulation on 7TLL.
Table 6 the results of molecular docking of Cissampeline compounds have hydrogen bonds at the amino acid residue of the 7TLL receptor, namely Phe:112. Meanwhile, the results of molecular dynamics simulations show that the Cissampeline compound has hydrogen bonds at the amino acid residues of the 7TLL receptor, namely Thr: 90, Thr: 24, and Glu: 166. The results of molecular docking of native ligand with the 7TLL receptor showed hydrogen bonds at residues Val:148, and Phe:150 while after molecular dynamics simulations showed that native ligand had hydrogen bonds at Gln:192, Glu:166, Hie:164, Hie:163, and Gly:143. Then, based on the results of molecular docking between Molnupiravir and the 7TLL receptor, no hydrogen bonds were shown. Meanwhile, based on the results of molecular dynamics simulations, it is shown that Molnupiravir has hydrogen bonds with 7TLL receptors on Glu166 and Gln192 residues. Cissampeline, a natural ligand, and Molnupiravir have hydrogen bonds at the same amino acid residue of the 7TLL receptor, namely Glu:166.
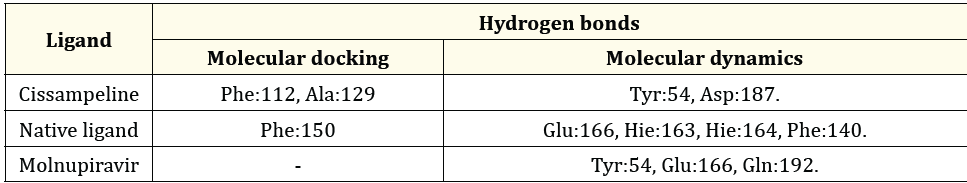
Table 7: The Comparison between hydrogen bonds formed during molecular docking and after molecular dynamics simulation on 7VH8.
Based on Table 7 the molecular docking results for Cissampeline compounds have hydrogen bonds at the amino acid residues of the 7VH8 receptor, namely Phe:112, and Ala:129. The results of the molecular dynamics simulation showed that the Cissampeline compound had hydrogen bonds at the amino acid residue of the 7VH8 receptor, namely Gln189. The results of molecular docking of native ligand with the 7VH8 receptor showed hydrogen bonds at the Phe:150 residue. In contrast, after molecular dynamics simulations showed that the native ligand had hydrogen bonds in Glu166, Hie:163, Hie:164, and Phe140. Then, no hydrogen bonds were shown based on the results of molecular docking between Molnupiravir and the 7VH8 receptor. Meanwhile, based on the results of molecular dynamics simulations, it shows that Molnupiravir has hydrogen bonds with the 7VH8 receptor at residues Tyr:54, Glu:166, Gln:192. Cissampeline compound has the same hydrogen bond as Molnupiravir at the amino acid residue of the 7VH8 receptor, namely at Tyr:54. And Molnupiravir has the same hydrogen bonding at the amino acid residue of the 7VH8 receptor as its natural ligand, Glu:166.
Cissampeline compound is a secondary metabolite of abuta (Cissampelos pareira Linn) which has good molecular docking score, and good prediction of ADMET. In addition, the compound complies with Lipinski's rule of five so that it can be used as a drug candidate that can be given orally. Therefore, predictions of synthetic pathways for these compounds were carried out to find new compounds or substances that might have activity as medicinal compounds to create new chemical products. The prediction of the Cissampline synthesis pathway was carried out using the Chemical.AI website. Based on Figure 9 to produce Pyrrolezanthine compound, it is done by mixing 1.8 g of D-Fructose (10 mmol) and 247 mg of Tyramine (2 mmol) by dissolving in 40 mL of acetic acid and 30 mL of triethylamine (TEA) and stirring at room temperature. 80oC for 7 hours. The sample was then diluted with 50 mL ethyl acetate and 50 mL H2O. The organic layer formed was then washed with NaHCO3 solution and dried with Na2SO4. The solvent was removed by vacuum, and then the residue was purified by silica gel chromatography giving a bright yellow color. Then the mixture was mixed with the compound 1-benzyloxy-4-(2-methane sulfonyloxyethyl) benzene and sodium hydride and dimethyl formamide at 60oC for 45 minutes to produce compound 1-[2-(4-{2-[4-(benzyloxy) phenyl]ethoxy} phenyl)ethyl] -5-(hydroxymethyl)-1H-pyrrole-2-carbaldehyde. The compound was then reacted with KBr to produce 99% Cissampline compound with 66% similarity.
The secondary metabolite of abuta (Cissampelos pareira Linn) which has the best docking score at the 5R7Y receptor is Isochondodendrine with a docking score of -94.60; on the 7JKV receptor was Cissampaline with a docking score of -94.87; at the 7TLL receptor were Kaempferol-3-O-β-D-Glucopyranoside with a docking score of -99.03; and 7VH8 is a Cissampline with a docking score of -99.03. Cissampline is one of the compounds that had good docking scores at 5R7Y, 7JKV, 7TLL, and 7VH8 receptors, met Lipinski's rule of five, had good absorption, distribution, and excretion values, and did not cause hepatotoxicity. The interaction between the Cissampline compound and the four receptors was also stable at the time of molecular dynamics simulation for 10 nanoseconds. The average result of the interaction between Cissampline and the 5R7Y receptor RMSD was 1.77Å; with the 7JKV receptor is 1.45Å; with 7TLL receptors is 1.59Å; and with 7VH8 receptors is 1.62Å. The average RMSF value of Cissampline compounds with 5R7Y receptors is 0.88Å; with the 7JKV receptor is 0.91Å; with 7TLL receptors is 0.93Å; and with 7VH8 receptors is 0.95Å. As a result, the Cissampeline compound appears to exhibit anti-COVID-19 activity.
There is no conflict of interest in this research.
Copyright: © 2024 Saeful Amin., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff







