Michael Chistodoulou1*, Kontogeorgakos Vasileios3, Flieger Ioannis4,Helen Fryssira2and Sophia Kitsiou5
1Orthopaedic Surgeon,University of Athens, Greece
2Department of Medical Genetics, Choremeion Research Laboratory, National and Kapodistrian University of Athens,Greece
31st Orthopaedic Clinic, Attikon University Hospital, National and Kapodistrian University of Athens,Greece
4Agia SophiaChildrens Hospital, 1st Orthopaedic clinic, Athens,Greece
5Retired Professor of Clinical Genetics, University of Athens, Greece
*Corresponding Author: Michael Chistodoulou, Orthopaedic Surgeon, University of Athens, Greece.
Received: February 23, 2021; Published: March 27, 2021;
A case of a novel in-frame FLNB deletion in familiar Larsen syndrome of a newborn, with a clinical appearance of dislocated bilateral hip joints, knee joints, elbows and equinovarus foot deformities. Patient underwent both conservative treatment and surgical treatment in sequence of severity as indicated, while in parallel genetic counseling with subsequent DNA testing revealed the familiar extent of the syndrome and a novel mutation first reported today. Patient follow up is presented until adulthood giving a great insight to the impact of the treatment chosen at presentation.
Keywords: Larsen Syndrome; Hip Dislocation; Knee Dislocation; Elbow Dislocation; Equinovarus Deformity; FLNB Deletion
Autosomal Dominant Larsen Syndrome (LS)(OMIM 150250) was originally described by Larsen and associates in 1950 [1]. The incidence is 1 in 100.000 live births. There is also a rare recessive Larsen syndrome (OMIM No.608637) which is due to mutation at CHST3 gene and the protein affected is the Carbohydrate sulfotransferase 3; chondroitin 6-sulfotransferase with more severe joint disease but without the characteristic flat face of dominant Larsen syndrome [2]. The autosomal dominant form being more common and many cases have at present an identifiable genetic etiology in the FLNB gene [3].
Patients born with LS syndrome have multiple orthopaedic defects. Mainly, dislocations of major joints hyperextension deformities of knees [4], shortened metacarpal bones and a spatulate thumb that could be dislocated at the metacarpal joint, scoliosis, equinovarus deformities and rarely cervical kyphosis [5]. Management of those multiple joint disorders is quite challenging for the orthopaedic surgeon. At facial bones, patient could exhibit cleft palate and has a characteristic depressed nasal bridge, widely spaced eyes, prominent forehead and flattened midface [6]. Other problems these children face are mainly cardiac anomalies such as mitral valve prolapse, mitral regurgitation, atrial septal defect, aortic dissection and patent ductus arteriosus [5].Larsen syndrome is due to a defect in the encoding protein Filamin B. Filamin B binds to another protein actin changing the shape of a cell allowing it to move. Filamin proteins are large cytoplasmic actin-binding proteins which act and regulate the cytoskeletal network [Stossel.,et al. 2001]. Filamin B is important to chondrocyte maturation and differentiation.
We describe a familial case of a female patient with the typical clinical phenotype of Larsen syndrome who had a novel in frame deletion in the FLNB gene and the surgical treatment which she underwent.
The patient is a female born in March 2003, the 2nd child of a 31-year old father and a 29 - year old mother. She was born after a gestational period of 38 weeks by elective cesarean delivery due to breech presentation. Gestational period was generally uncomplicated with a single minor and brief incident of urethral bleeding.
At birth fetus had a normal Apgar score, weight of 3.500 gr, height of 50 cm and head circumference of 35 cm.
(Figure 1)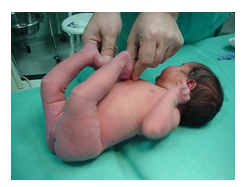
Figure 1:Genu recurvatum immediately after birth.
At birth, orthopaedic examination of the fetus presented with apparent bilateral knee dislocation in a position of hyperextension with a -40 degree angle and equinovarus position of bilateral feet.
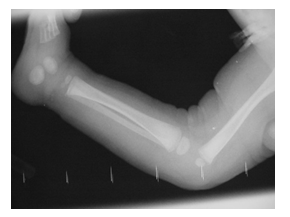
Figure 2:Grade 1 dislocation of the knees in Xray.
Radiological examination revealed a grade 1 dislocation of both knees, there were no signs of cervical kyphosis. Ultrasound examination of both hips revealed hip dislocation bilateral. There was a suspicion of possible bilateral radial head subluxation. The patient was evaluated by a Clinical geneticist and the diagnosis of Larsen syndrome was suspected.
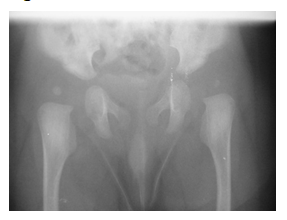
Figure 3:Dislocated hips bilateral on X-ray.
Patient’s family history indicates that her father was treated for bilateral hip dislocation diagnosed at birth by surgical reduction and also underwent surgical treatment of an equinovarus malformation of both feet.
Conservative knee reduction treatment was initiated to the patient immediately after birth with sequential cast applications, by use of tibial traction and flexion under mild sedation in order to avoid hinge subluxation at flexion.
In approximately 20 days of treatment, patient had acquired 35 degrees of knee flexion in a cast with adequate opposition of articular surfaces of femur and tibia, hence without any sign of subluxation, with only a slight proximal tibial bowing as flexion reached progressively to 90 degrees.
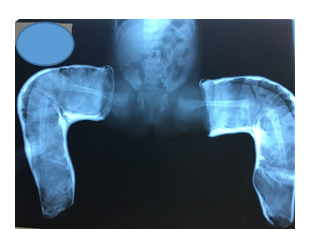
Figure 4:Cast treatment for dislocated knees including initial steps of the Ponseti technique to treat the clubfoot while in abduction to keep the hips centered within the acetabulum.
Progression of knee flexion led to initiation of closed reduction treatment of bilateral hip dislocation by use of hip spica placed under sedation in the safe zone of flexion-abduction.
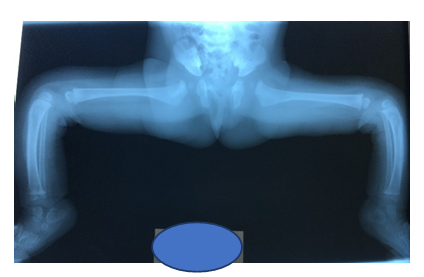
Figure 5:Note the correction of the knees followed by a tibial bowing usually self-limited as child grows.
By age of 6 months due to persistence of a left hip subluxation, open left hip reduction via a Smith Peterson approach was decided. Surgeon felt that there was no need for any femoral shortening as knee flexion was responding well to treatment.
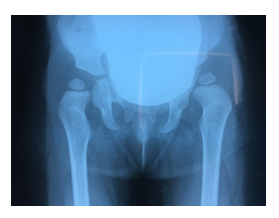
Figure 6: Failed conservative treatment of hip reduction note the insufficient acetabular index especially to left.
Open reduction, capsulorrhaphy and psoas tenotomy were performed, and patient was placed in a spica cast.
Patient started walking by age of 16 months. She had mild facial dysmorphism with mid face hypoplasia depressed nasal bridge and high palate. Her walking habitus was normal without any signs of Trendelenburg gait. X-ray checkup showed well centered hips without any signs of epiphyseal delay in growth as a result of the abduction position or the surgical approach, which would otherwise constitute reasonable reasons for epiphyseal ischemia either mechanical in the case of the abduction position or traumatic in the case of the surgical reduction.
Conservative treatment of bilateral feet equinovarus was initiated with use of sequential cast application following the Ponseti principles. Because of a persistent varus metatarsus, that was unacceptable by patient’s parents, she underwent surgical correction by age of 3.
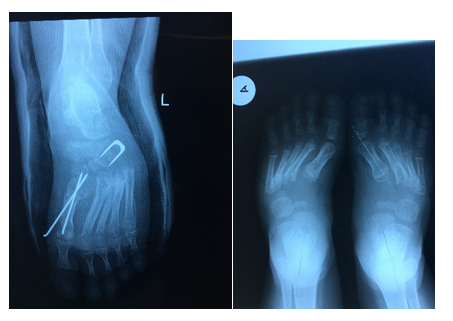
Figure 7:Pre and Post-surgical images of surgical correction of persistent clubfoot.
By the age of 15 patient is fully active, with a minor restriction of 10 degrees of supination and 5 degrees of pronation, due to bilateral radial head congenital dislocation. There was no evidence of hearing defect. Echocardiography and Ophthalmology examination were normal. Radial head protrusion in the elbow joint is not yet neither a cosmetic, nor a major functional concern. The presence of a persistent mild “bean shaped” foot (varus of metatarsals) bilateral doesn’t pose any difficulty to patients’ everyday activities. Longer follow up is necessary to ensure maintenance of correction.
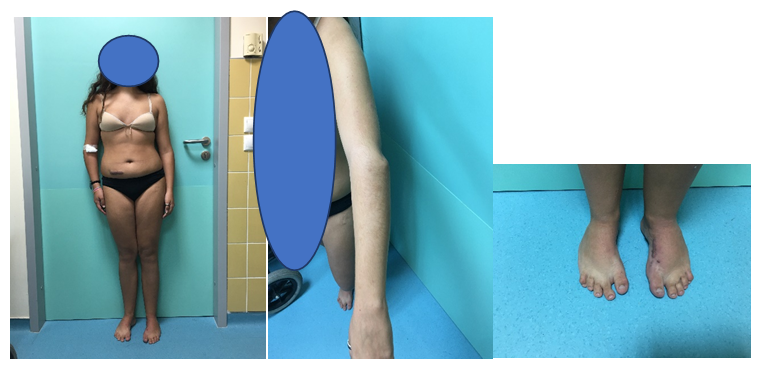
Figure 8:Teenage appearance, note the symmetrical stance stature, the clinical appearance of the radial head dislocation and the bean shaped feet.
After an informed consent from the patient family extraction of DNA from peripheral blood samples, from the patient and her father were obtained.
Approximately 37Mb (214,405 exons) of the Consensus Coding Sequences (CCS) were enriched from fragmented genomic DNA by an illumina platformto an average coverage depth 70-100X. An end to the end inhouse bioinformatics pipelines including base calling, primary filtering of low quality reads and probable artefacts, and annotation of variants was applied.
All disease causing variants reported in HGMD in ClinVar or in CentoMD (class 1) as well as all variants with minor allele frequency (MAF) of less than 1% in ExAc database were considered. Evaluation was focused on exons and intron boundaries +/-20. All relevant inheritance patterns were considered and provided family history and clinical information were used to evaluate eventually identified variants. Any relevant variant identified by NGS are in-house validated or Sanger sequenced in forward and reverse direction to exclude NGS artefacts.
The DNA analysis revealed that patient, had an FLNB heterozygous gene variant that consisted of an in framec.5117_5119 deletion expressing the protein p.(Phe1706del), which compared to existing lists of mutations has never before been reported.
Sanger sequencing: Exon 30 of the FLNB gene was analyzed by PCR and sequencing of both DNA strands of the entire coding region and the highly conserved exon-intron splice junctions. The reference sequence is NM_001164317.1.
It is categorized as class 3, hence of uncertain significance (VUS) according to the recommendations of ACMG, as it was in only our patient described so far. Subsequently, in the affected father was also confirmed the same variant in FLNB and a re-classification of the variant was done.
So, this novel in frame deletion is the cause of the patient’s and her father disease due to their typical clinical phenotype, radiological and familial presentation.
Larsen.,et al.in 1950[1], described multiple congenital large joint dislocations with characteristic facial anomalies in six independent patients.The syndrome is genetically and molecularly heterogeneous. The autosomal dominant form has been associated with mutations in the FLNB a cytoskeletal structural protein which probable play a role in vertebral segmentation, joint formation and skeletogenesis [Krakow.,et al. 2004]. Mutations in FLNB are exclusively associated with skeletal abnormalities showing a high histological specificity of FLNBmutations pathogenesis to the skeletal system. The phenotypic analysis of both carriers (patient and her father) confirms clinical and radiological signs of Larsen’s syndrome of different severity which can be remarkable due to intrafamilial genetic heterogeneity. This coincides with the intrafamilial variability described by Bicknell.,et al[7].
Closest mutation by deletion in relation to the above mentioned, was announced by Daniel.,et al. [8] which was a c.5023_5025delexpressing p.(Phe1675del)of the FLNB gene as it being a new mutation never before reported. Hickey.,et al. [9], also was found a heterozygous 3-bp deletion in FLNB in a child and her mother with mild clinical features consistent with Larsen syndrome.FLNB - related disorders include a spectrum of phenotypes ranging from mild (spondylocarpotarsal synostosis and Larsen syndrome) to severe (atelosteogenesis types 1 and 3 and Boomerang dysplasia) [10]. Spondylocarpotarsal synostosis (STS) (OMIM 272460) is associated with homozygous or compound heterozygous loss-of-function variants characterized by disproportionate short stature skeletal dysplasia with carpal and tarsal synostosis. The genotype-phenotype and the pathophysiology of abnormal FLNB function in Larsen syndrome remains unclear [9]. Variants detected in Larsen syndrome are mostly missense variants orless commonly, as in our patient, in frame deletions.
Identification of causative variants is important for the management of patients. Significant progress in the understanding of the molecular defects underlying a spectrum of skeletal malformations including Larsen syndrome has been achieved with the next generation sequencing technology. NGS is currently considered a high throughput method for detecting rare variants in genetically heterogeneous diseases [11].Has certainly proved cost- effective for the study of substantially large genomic regions, as well as the synchronous analysis of more than one genetic loci. The undiagnosed patients with mild hypoplasia of the skeletal system may need to proceed to whole genome sequencing or other methods to allow identification of the underlying mechanism.
Hence the clinical diagnosis of Larsen’s syndrome was confirmed giving the opportunity for the patient to use this information for future early prenatal diagnosis thus, stopping the chain of the genes inheritanceby choice.
The clinical appearance of such a baby causes a great challenge to any surgeon. A herculean task [12] as it is described in literature. The sequence in treatment priority was followed as it is described by Tachdjian’s Pediatric Orthopaedics textbook, and the results proved the accuracy of the guide. Uncertainties still remain as to the best approach to the foot deformities as those persist due to the soft tissue laxity, but the surgeon’s preference proved to provide to this age a functional painless foot[13,14].
Citation: Christodoulou Michael., et al. “Novel In-Frame FLNB Deletion in Familiar Larsen Syndrome. A Case Report".Acta Scientific Orthopaedics 4.4 (2021):68-73.
Copyright: © 2021 Christodoulou Michael., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.















 +91 9182824667
+91 9182824667
 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff
