Carlos A Guerrero*
Department of Physiological Sciences, Faculty of Medicine, Universidad Nacional de Colombia, Bogota, Capital District, Colombia
*Corresponding Author: Carlos A Guerrero, Department of Physiological Sciences, Faculty of Medicine, Universidad Nacional de Colombia, Bogota, Capital District, Colombia.
Received: April 09, 2021; Published: May 17, 2021
Citation: Carlos A Guerrero. “Generate Intracellular Oxidative Stress is an Obligatory Mechanism of RNA Viruses During the Infectious Process”. Acta Scientific Microbiology 4.6 (2021): 70-81.
Viruses, upon contact with the host cell, make conformational changes in their capsid proteins, leading to the emergence of molecular domains needed for binding to cell receptors in order to facilitate penetration of the virus into the cell. Generally, these conformational changes require the participation of molecules with redox capacity in the cell membrane. Once viruses enter, they need to induce conditions of cellular stress, usually oxidative stress, rapidly. This is a necessary condition for the cell to express molecular factors necessary for the viral RNA to be translated into the corresponding proteins that will allow a successful viral infection. In parallel, or as a consequence of the same oxidative process, the virus induces pro-inflammatory pathways such as NF-kB and concomitantly inhibits anti-inflammatory pathways as superoxide dismutase, Nrf2, and PPARγ. Likewise, the viral infection maintains the oxidative condition because it allows the assembly of virions by facilitating the native conformation of their proteins in which the formation of intermolecular disulfide bridges occurs. Under these conditions, the administration of drugs or antioxidant molecules interferes with the virus's oxidative conditions for its entry, replication, and assembly of new virions. In this way, the amount of virions generated per infected cell is drastically reduced, giving the immune system the opportunity to control the infection or reduce its aggressiveness.
Keywords: IRES; NF-κB; ROS; Oxidative Stress; Antioxidants
EPA: Eicosapentaenoic Acid; DHA: Docosahexaenoic Acid; RA: Retinoic Acid; PGZ: Pioglitazone Hydrochloride; RGZ: Rosiglitazone Maleate; NAC: N-Acetyl Cysteine; AA: Ascorbic Acid; IKK: IκB Kinase; IκB: Nuclear Factor of Kappa Light Polypeptide Gene Enhancer in B-cells Inhibitor; NF-κB: Nuclear Factor Kappa-light-chain-enhancer of Activated B Cells; PPARγ: Peroxisome Proliferator-activated Receptor Gamma; NSAIDs: Non-Steroidal Anti-inflammatory Drugs; IBF: Ibuprofen; ASA: Acetylsalicylic Acid; COX-2: Cyclooxygenase-2; PDI: Protein Disulfide Isomerase; RXR: Retinoid X Receptor; ILs: Interleukins; TNF: Tumor Necrosis Factor; IFN: Interferon; ROS: Reactive Oxygen Species; TRIF: TIR-domain-containing Adapter-inducing Interferon-β; TLR: Toll-like Receptor; NADPH Oxidases (NOX) Enzymes
Some viruses that affect animals, including humans, such as chikungunya, dengue, coronavirus (including SARS-CoV-2), or plants, share similar infection mechanisms [1-5]. It has been found that most RNA viruses induce oxidative stress in the host cell and that the virus can use this mechanism associated with the defense response to increase replication and modify the cellular biochemistry for its own benefit. It is known that respiratory viruses induce ROS-generating enzymes, including nicotinamide adenine dinucleotide phosphate oxidases (NADPH oxidases, Nox) and xanthine oxidase (XO) that also decrease antioxidant compounds [2]. Also, it has been shown that conditions that alter the cellular redox balance, either by antioxidant deficit or increase of free radicals, can contribute to viral pathogenicity [2,5]. On the other hand, it is known that oxidative stress is a response to the alteration of homeostasis during viral infection and maybe a mechanism by which viruses cause cellular and tissue damage. Canonical signaling of NF-κB, which regulates the innate immune response, cell survival, and inflammation, is manipulated by viruses to counteract antiviral response. It has been observed that some viruses modify NF-κB expression to alter the cellular mechanisms that eliminate infection is also known that ROS activates signaling pathways such as NF-κB or vice versa [6,7]. Our research group has reported that during rotavirus infection, the expression of cellular proteins such as NF-κB, Cox-2, Hsc70, PDI, and PPARγ increases. When cells are treated with PPARγ agonist, pioglitazone, the expression of these proteins decreases, and there is a reduction in the production of virions [8]. Likewise, by interfering with oxide reduction activity by applying antioxidants or applying non-steroidal anti-inflammatory drugs (NSAIDs) to decrease NF-κB pathway activity, rotavirus infection is decreased in the MA104 cell line [9], in isolated mouse villi [8], in ICR mice [10]and children [11]. NF-κB activation favors herpes simplex virus infection (HSV), hepatitis C virus (HCV), poxvirus, and cytokine induction [2]. The current pandemic virus, SARS-CoV-2, positively regulates the NF-κB signaling pathway [3]. The excessive inflammatory response to respiratory viruses is largely due to deregulation in the immune system manifested as secretion of pro-inflammatory cytokines, activated by NF-κB [6]. The relationship between oxidative stress and viral infection is not fully known, as are the mechanisms by which its effects on cellular functions lead to cell death.
The purpose of this review is to contrast the results of research conducted on rotavirus by the rotavirus research group of the Medicine School of the National University of Colombia and other groups with what has been found in the literature related to the question ¿Why do RNA viruses generate intracellular oxidative stress as an obligatory mechanism during the infectious process?
The translation of viral RNA is dependent on IRES.The translation is the process by which a protein is synthesized from the information contained in a molecule of messenger RNA (mRNA) in the ribosomal complex. However, when cells enter a stressful condition, they modify the ribosomes' utilization in mRNA translation, changing the 5'cap dependent mechanism to a 5'cap independent mechanism [12-14]; likewise, it is an initial mechanism host self-defense against viral invasion. Blocking of 5'cap translation is observed in most cellular stress conditions, including oxidative stress, viral infection, nutrient limitation, temperature changes, hypoxia, ultraviolet irradiation, endoplasmic reticulum stress, among others. To adapt to these stress conditions, both the cell and the virus need to adjust their translation initiation mode. Many viral infections trigger endoplasmic reticulum (ER) stress responses in various ways within the host cell. One way to inhibit mRNA translation is by blocking 5'-cap-dependent translation. The second form of translation inhibition is due to the phosphorylation of eIF2α. Four kinases respond to stress and can affect overall translation by phosphorylation of eIF2α, which are: heme-regulated inhibitor kinase (HRI), protein kinase RNA (PKR), PKR-like endoplasmic reticulum kinase (PERK), and general control nonderepressible 2 (GCN2). These kinases are activated by different stress types that induce a common pathway for blocking translation. There are similarities in the translation control strategies in virus-infected cells and uninfected cells with stress. For example, under conditions of cellular stress or in virus infection, internal ribosome entry sites (IRES) allow mRNA translation because 5'cap dependent translation, mediated by eIF2α and by eIF4F, is inhibited [13]. IRES are regulatory elements of mRNA translation that were first discovered in viruses and can be read by cell biochemistry expressed in stress. This biochemistry can vary depending on the virus. Another similarity is that in cells under stress or infected by viruses, the phosphatases are used to prevent the accumulation of phosphorylated eIF2α since this stops the synthesis of proteins. Similarly, the eIF4G factor can be cut by proteases either encoded by viruses or by the cell during apoptosis. For example, picornaviruses express a protease that cuts the eIF4G initiation factor, a component of the 5'cap binding complex, blocking the 5'cap dependent initiation process by stopping translation in the infected cell. Some stress-related mRNAs from mammalian cells have IRES similar to that of viral RNAs [14]. Previously it was thought that IRES could not translate eukaryotic mRNAs and that the only mechanism was the 5'cap dependent process involving recruitment of the initiating subunits and the 40 S at the 5' end of mRNA. These cellular IRES elements are located at the 5'UTR end (untranslated region) of the eukaryotic mRNA genes involved in survival to cellular stress and other critical survival processes. It is thought that IRES allows the translation of cellular mRNA when the 5' cap-dependent process is blocked, as occurs during mitosis (G2-M phase) and under stress conditions such as heat shock or viral infection. For example, during apoptosis, the X-linked apoptosis inhibitor mRNA (XIAP) containing IRES, apoptotic peptidase activating factor 1 (APAF1), and p53 are expressed. IRES has also been found in the proto-oncogene c-myc, fibroblast growth factors (FGFs), vascular endothelial growth factors (VEGFs), and master regulator genes of cellular responses [15]. The p53-related IRES are induced when there is stress by genotoxic or cytotoxic agents as well as the mRNAs IRES of heat shock Proteins, such as BIP protein, heat shock protein 70 (Hsp70), Hsp22, and Hsp27 [15].
RNA or DNA viruses recruit the cellular machinery to translate viral mRNA. RNA viruses require inducing oxidative stress to generate changes in redox homeostasis so that the biochemical machinery needed to translate their RNA and synthesize viral proteins is expressed. These viruses use IRES to keep viral translation active when host cell translation is inhibited. It is known that in human immunodeficiency virus 1 (HIV-1), oxidative stress is caused during infection, which stimulates IRES activity in infected cells. Various viruses induce oxidative stress to facilitate replication within the cell [2,17]. Thus, oxidative stress induced by the infection activates several critical antivirals signaling pathways such as the Toll-type receptor (TLR) and interferon pathways (IFN). It has been shown that increased oxidative stress in a host cell contributes to viral pathogenesis, resulting in a massive induction of cell death. Basically, the imbalance between ROS production and the antioxidant defense system directly relates to the pathogenic mechanisms for the inflammatory response and tissue injury by a viral infection. The increase of oxidative stress is partly a consequence of the activation of the NF-κB pathway and the blocking of cellular proteins' normal translation directly by viral proteins or interferons. These host translation inhibition mechanisms are diverse and can be initiated either by the virus or by the cell itself, depending on the virus type [2,5]. Although initially, the IRES were discovered in picornavirus, poliovirus (PV), and encephalomyocarditis virus (EMCV), to date, 60 human and animal viruses and eight plant viruses have been reported to contain IRES elements. Many viruses have IRES in their RNA, such as Picornaviridae, Flaviviridae, Dicistroviridae, Retroviridae and coronaviridae, including SARS-CoV-2 [13,18]. SARS-CoV-2 NSP1, which was proposed to be a virulence factor that inhibits protein synthesis by directly binding the human ribosome [19].
Viral RNA IRES has complex conformations and is only active when bound to specific proteins called IRES transactivation factors (ITAFs). ITAFs do not work in 5'cap dependent translation and are all of cellular, non-viral origin; they are important, as IRES must work before any viral protein can be synthesized. From the moment the virus enters the cell, it must induce oxidative stress to activate the biochemical machinery to stop the 5'cap dependent translation and activate ITAFs. In both stressed cells and viral infections, IRES plus required ITAFs and the canonical translation factors work together as a complex. It is believed that many of the ITAFs in their interaction with the IRES have similar functions, both for cellular mRNAs and viral mRNAs, during 5'cap independent translation [13,15]. This suggests that ITAFs themselves can control both viral and cellular IRES, probably because they act with a similar interaction mechanism with cellular or viral IRES, or the same ITAF may have different mechanisms that allow it to interact with viral or cellular IRES. When cellular or viral IRES are analyzed using fibroblast growth factor 2 (FGF-2 IRES) vectors, these IRES are active in most types of transiently transfected human and non-human cells. In vivo, in mice, FGF-2 IRES activity is low in most adult organs but exceptionally high in the brain. This space-time variation is not observed in the IRES of viral origin, indicating tissue specificity of cellular IRES but not viral IRES [13]. The same ITAF can regulate different cellular IRES, and a given IRES can be regulated positively or negatively by several ITAFs. Without cellular stress, IRES activity is inhibited by negative ITAFs, while under stress, the positive ITAFs that bind to IRES increase, which facilitates the unwinding of the RNA's secondary structure, improving IRES activity. IRES is commonly found in 5' RNA UTR of RNA viruses and allow RNA translation independently of 5' cap. However, DNA viruses, including simian virus 40 (SV40) and herpesvirus, also contain polycistronic mRNAs with IRES. The best-studied DNA viral IRES is that of Kaposi's sarcoma herpesvirus (KSHV) and Marek's disease virus [20].
During cellular stress without viral infection, or in stress by infection, cellular mRNAs containing IRES responsible for cell survival/growth, such as BiP, Bcl-2, and VEGF, are translated by the IRES-dependent mechanism using ITAF. This allows cells to respond quickly to transient changes and delay apoptosis. If the stress condition is removed, the cells resume normal activity. However, if the stress is prolonged or severe, as in persistent viral infection, the pro-death genes (Apaf-1, DAP5, CHOP, p53, etc.) are also selectively translated by the same IRES mechanism, which allows cells to adjust their response to cellular stress. If stress conditions do not stop and cellular homeostasis is not restored, these proteins induce apoptosis [13].
Another evidence that indicates that viruses require inducing oxidative stress to succeed in the infectious process is IRES increased activity when cells are treated with agents that induce oxidative stress. Flaviviridae viruses such as Hepatitis C virus (HCV), Dengue virus (DENV), Zika virus (ZIKV), Japanese encephalitis virus (JEV), West Nile virus (WNV), and Tick-borne encephalitis virus (TBEV) induce and enhance their infective capacity with oxidative stress. At the same time, the oxidative stress induced by the viruses explains their pathogenesis [2]. Most flavivirus-gender viruses have in the 5' UTR a methylated 5'cap (type I cap (m7GpppNmN), while other gender viruses possess IRES. However, HCV is known to use IRES in translation, which allows the selective translation of viral genes when the synthesis of global host proteins has been decreased. HCV translation is increased when treating cells with H2O2, suggesting that HCV can adapt and use the host's antiviral response to facilitate its own translation, allowing the virus to perform the full infectious cycle under oxidative stress conditions, maintaining chronic infectivity. Additionally, our results [5,8] indicate that rotavirus infection in intestinal cells increases ROS generation. An increase in virions formation is also observed when infected cells are treated with ROS generating drugs such as H2O2, drugs used in chemotherapy (cisplatin, doxorubicin), metronidazole, or nitrofurantoin (unpublished data).
Activation of the NF-κB pro-inflammatory pathway and inhibition of anti-inflammatory pathways such as PPARWith rotavirus, as with coronavirus [3,5,21,22], the infection increases oxidative stress and induces an imbalance in reduced and oxidized glutathione levels, indicating an early alteration of the intracellular redox state. Our research group has also found that when increasing the oxidative stress in the cell and infect it, the percentage of infection and the number of virions is increased. For example, in an in vitro model, when MA104 cells are treated for 1 hour with agents 2-3-Dimethoxy-1,4-naphthoquinone (DMNQ) and homocysteine, which increase ROS levels and the activation of the NF-κB pathway, and then we perform the infection with RRV rotavirus, ROS levels increase between 2.1 and 2.4 times and infection levels increase between 20% and 30% respectively (unpublished data). This suggests that an increase in oxidative stress and activity of the NF-κB pathway is associated with an increase in the percentage of viral infection. A virus needs to express different proteins within a host cell, both structural and non-structural, to replicate the viral genome and assemble a new virion. It has been shown that different viral proteins are related to the production of several ROS [17]. Virus-induced oxidative stress plays a central role not only in the successful completion of the viral life cycle but also in viral pathogenesis [22]. In the influenza virus, among other respiratory viruses, and Sindbis virus (an alphavirus with RNA positive genome with envelope), the NF-κB pathway's activity contributes to virus replication and spread. The increase of oxidative stress as the increase of the NF-κB pathway activity induces the infection's increase. The above reinforces the hypothesis that an oxidized state's imbalance promotes a cellular environment conducive to the viral infection's success, as observed in our rotavirus results [8].
It has been proposed that in patients with SARS-CoV-2, oxidative stress is associated with amplification and maintenance of the cytokine storm, coagulopathy, and cellular hypoxia [21]. The inflammatory process is an important element in the cytokine storm [23], and studies indicate that ROS activates the pro-inflammatory factor called P3 NOD-like receptor (NLRP3). The activation of oxidative stress by viral infection activates Toll-type receptor and interferon (IFN) pathways as a mechanism to fight viral infection. NF-κB, in turn, is activated by ROS, so directly or indirectly, the inflammatory process is increased by ROS [24]. High levels of sterol oxidation products have been detected during the infection and up to three months after not having the viral infection. Despite the antiviral function of cytokines in respiratory infections, their excessive production during the cytokine storm is more damaging to lung tissues than the viruses themselves. On the other hand, ROS-induced cell death and lysis may favor the release and dissemination of virions and stimulate replicating those respiratory viruses with a lytic life cycle. Therefore, several agents have been evaluated to control the infection and viral spread, and the inflammation associated with the infection. Much attention has been paid to antioxidants because of the correlation between the severity of tissue injury and oxidative stress markers in infected patients' lungs and blood, such as with the human respiratory syncytial virus (HRSV) [6].
N-acetylcysteine (NAC) is known to lower the viral load by increasing cell redox status, maximizing glutathione synthesis, and potentially decreasing virus-induced oxidative stress and cell death [25]. NAC has also been shown to inhibit the inflammatory process by decreasing NLRP3 (IL1β and IL18) in vitro and to reduce plasma TNF-ɑ, pro-inflammatory cytokines (IL-8, CXCL10, CCL5, and IL-6) in human clinical trials [26].
Oxidative stress generated by viral infection plays a key role in activating innate immunity by generating cytokines when the NF-κB pathway is activated. Respiratory syncytial virus (RSV) induces ROS production, which, in turn, induces the expression of pro-inflammatory cytokines and innate immune defense. To control ROS levels, viruses have evolved to manipulate anti-inflammatory pathways, such as the Nrf2 pathway [17]. Nrf2 is a protein that detects oxidative stress levels and regulates the genes involved in producing antioxidant enzymes and stress response genes. During viral infection, the cellular antioxidant response is inhibited by increasing the degradation of Nrf2 protein, which correlates with an increase in viral antigens [17]. For example, RSV increases lipid peroxidation and decreases GSH in human type II alveolar epithelial cells and small airway epithelial cells and reverses Nrf2 pathway activation, causing a reduction in Nrf2 target genes expression, including hemoxigenase-1 (HO-1), superoxide dismutase 1 (SOD1), superoxide dismutase 3 (SOD3), glutathione S-transferase (GST), catalase (CAT) and glutathione peroxidase (GPx). On the other hand, it has been shown that an alteration in the redox balance of a host cell contributes to viral pathogenesis, resulting in a massive induction of cell death by oxidative stress [2]. However, studies show that examples of positive modulation of the Nrf2 pathway by virus-induced oxidative stress prevent early cell death [17]. Influenza viruses induce oxidative stress and facilitate nuclear translocation of Nrf2 with subsequent expression of HO-1, a protective enzyme against oxidative injury in human alveolar epithelial cells. This virus finally induces apoptosis and cytotoxicity in alveolar epithelial cells along with an increased expression of caspase 1 and 3 and a pro-inflammatory cytokine, IL-8. These findings support the idea that oxidative stress is significantly involved in the success or failure of the cellular response to virus infection.
On the other hand, the peroxisome proliferator-activated receptors (PPAR) play an important role in the antagonism of central inflammatory pathways such as NFκB, AP1, and STAT. PPARα and PPARγ regulate the inflammatory response in vivo and in vitro by inhibiting the activation of NFκB. PPARs activate the transcription of target genes by forming heterodimers with the 9-cis retinoic acid receptor (RXR). The complex binds to specific sequences called peroxisomal proliferator response elements (PPRE) located in the target genes promoter, which regulate the transcription of these genes. In infection by RRV rotavirus, we have observed a decrease in the expression of the PPRE response element in infected MA104 cells, with respect to uninfected cells, suggesting that RRV inhibits the anti-inflammatory pathway by decreasing the transcriptional activity of PPRE (unpublished data). Influenza A virus (IAV) negatively regulates PPARγ after alveolar macrophage (AM) infection through interferon type I dependent signaling (IFN). Similarly, PPARγ expression in AM suppresses the exaggerated antiviral and inflammatory AM response after IAV and respiratory syncytial virus (RSV) infections. PPARγ deficiency in myeloid cells resulted in increased host morbidity and increased lung inflammation, suggesting that PPARγ macrophage is vital in restricting IAV and RSV development in the patient [27]. Also, it has been observed that the treatment with rosiglitazone (RGZ) induces a significant decrease of the human immunodeficiency virus HIV-1 in macrophages; the stimulation of PPARγ with RGZ inhibits the virus replication modulating the NF-κB activation, which leads to the negative regulation of the long terminal repeat promoter activity (LTR) of HIV-1 and the suppression of HIV-1 replication. Also, decreased influenza viral load and decreased cytokine and chemokine production have been found in rosiglitazone-treated, influenza-infected mice compared to non-infected mice [28]. PPARγ agonists positively affect the suppression of the inflammatory response during respiratory syncytial virus (RSV) infection. All of this indicates that PPARγ activation is an effective therapeutic strategy for countering the cytokine storm and preventing inflammatory effects that follow coronavirus or other RNA virus infections. PPARγ agonists have anti-inflammatory properties that make them promising candidates for treating inflammation in severe viral diseases. Natural PPARγ agonists such as 15d-PGJ2 and synthetic ones such as thiazolidinediones significantly downregulate ICAM-1 expression induced by the respiratory syncytial virus (RSV) in A549 and NHBE cells.
Different investigations have reported that PPAR𝛾 activation by thiazolidinediones (TZD) such as rosiglitazone (RGZ), pioglitazone (PGZ) or by flavonoids, fibrates (fenofibrate, gemfibrozil) [29] interferes with the NF-κB signaling cascade, which leads to a decrease in the transcription of some NF-κB dependent pro-inflammatory genes and the inhibition of viral infections or to the modification of the inflammatory process caused by the viral infection. When the NF-κB pathway is activated, pro-inflammatory COX-2 expression is activated; PPARγ ligands exert their chemoprophylactic properties by preventing p65/p50 translocation nucleus, thus inhibiting NF-κB action and then Cox-2. Part of PPAR's anti-inflammatory regulation is mediated because PPAR negatively interferes with nuclear factors such as NF-κB, AP-1, and C/EBP, which regulate innate and adaptive immunity. PPAR𝛾 ligands inhibit the expression of inflammatory genes such as interleukin-1𝛽 (IL-1𝛽) and tumor necrosis factor-𝛼 (TNF-𝛼). PPAR𝛾, as well as PPAR𝛼 and PPAR𝛽, is expressed in a wide variety of cells in which it regulates the transcription of different genes through heterodimerization with retinoid X receptors (RXR). For this reason, vitamin A (holo-trans-retinol) also exerts anti-inflammatory effects. Patients with HIV infection have decreased expression of the PPARγ gene and proteins in alveolar macrophages, which correlates with increased oxidative stress. Treatment with a PPARγ agonist increases PPARγ levels, improves the phagocytic index of macrophages, and decreases oxidative stress, supporting the potential role of PPARγ agonists in treating viral infections.
Rotavirus infection induces an inflammatory response in the host cell accompanied by increased expression or activation of some cell molecules, including ROS, NF-κB, and COX-2 [8]. Our research group has reported that stimulation with PPAR𝛾 agonists (TZD, RGZ, PGZ, DHA, and ALA), all-trans retinoic acid (ATRA), and treatment with N-acetylcysteine (NAC) interfere with rotavirus infection, as has been reported for other viral infections [5,8-11]. The accumulation of pro-inflammatory cellular proteins and the increase of ROS, induced by rotavirus infection, is reduced with pioglitazone treatment, also producing a concomitant reduction in the yield of the number of infectious virions suggesting that PPARγ inhibits NF-κB [30]. Like many other RNA viruses, rotaviruses activate the inflammatory pathway, increasing NF-κB expression and inhibiting anti-inflammatory pathways such as PPAR [5,8]. Our research group has discovered that PPARγ is phosphorylated and not translocated to the nucleus during rotavirus infection, thus inactivating this anti-inflammatory pathway (unpublished data). Phosphorylation is the most important post-translational modification affecting PPARγ activity. PPARγ phosphorylation by MAPK, including p38MAPK, Jun N-terminal kinase (JNK), and extracellular signal-regulated kinase (ERK), causes ligand-dependent inhibition and ligand-independent protein transactivation. AMPK has also been shown to phosphorylate and inhibit PPARγ activity.
Why do antioxidants reduce virus generation, both in vitro and in vivo? ROS reduction and inhibition of pro-inflammatory pathway NF-κBOur research group has reported that rotavirus infection [5,8-11], like that of other viruses [6,31], decreases significantly with non-steroidal anti-inflammatory drugs (NSAIDs, ibuprofen, diclofenac), with the antioxidant NAC, PPARγ agonists (rosiglitazone and pioglitazone), polyunsaturated fatty acids (DHA), trans-retinoic acid (vitamin A), ascorbic acid, curcumin, both in vitro and in vivo, due to their effect as NF-κB-pathway inhibitors. When using these compounds, a decrease in rotavirus infection with a decrease in the expression levels of NF-κB, PDI, Hsc70, PPARγ, COX-2, and ROS has been generally observed, which reaffirms the importance of inflammatory status in rotavirus infection, as it has been reported for other viruses [2,21], suggesting that the antiviral effect of these substances is associated to the inhibition of these pathways. Since oxidative stress is the link of all known mechanisms for viral infection, including SARS-CoV-2, antioxidants, such as NAC and glutathione (GSH), donors of the SH group may have a positive effect on the recovery of patients with viral infections. Other compounds such as vitamin E, vitamins C, D, and selenium, chelating agents that form complexes with iron (deferoxamine), NF-κB inhibitors (NSAIDs, dexamethasone, curcumin, gingerol, etc.), polyphenols, Nrf2 activators (curcumin, resveratrol), and the use of specific pro-inflammatory cytokine inhibitors and anticoagulants [3,4,17,21,25,32-36] can also be used in the management of viral infections. NF-κB inhibitor drugs such as caffeic acid phenethyl ester (CAPE), Bay11-7082, and parthenolide that inhibit NF-κB activation have been reduced SARS-CoV-2 inflammation in mice. Synthetic triterpenoids (oltipraz, butylated hydroxyanisole), sulforaphane in the cabbage family (broccoli, Brussels sprouts, cabbage, cauliflower, and broccoli kale), curcumin in turmeric, and silibinin, the main active constituent of silymarin (Silybum marianum or sea thistle), resveratrol (present in red wine, red grape skin, blackberries) exerts antioxidant action that activates Nrf2 and inhibits NF-κB [37,38] pathways. The action of these agents down-regulates pro-inflammatory cytokines such as interleukin (IL)-1, IL-6, IL -8, and tumor necrosis factor (TNF-α). The results obtained in these studies suggest that decreasing oxidative stress is a viable strategy in the management of viral infections. By modulating redox-sensitive pathways, the immune response can be regulated and viral infection controlled, which has been studied in the context of a wide variety of viruses, including SARS-CoV-2 [3,5,21,25,32,34].
Glutathione (GSH) is the main antioxidant agent in mammals and is the most important antioxidant defense in the lungs. Normally, when the balance between GSH/GSSG is disrupted by an increase in ROS, a more oxidative environment oxidizes the proteins that control the activation and localization of transcription factors, such as KEAP-1, which regulates Nrf2, and IκB, which regulates NF-κB, activating the anti-inflammatory pathway and inactivating the inflammatory one [39]. In this way, the cell controls oxidative stress; however, in viral infections, KEAP-1 remains bound to Nrf2, causing its degradation via ubiquitination in the proteasome, or degrading its noncanonical pathways independent of KEAP-1 [40]. Coronavirus infections lead to alterations in the redox balance of infected cells through the modulation of NAD+ biosynthesis. Therefore, SH group donor molecules, Nrf2 activators, and NF-κB inhibitors are potential therapeutic options for RNA virus infection, including SARS-CoV-2 [34,39,41]. This therapeutic approach has been reported to reverse the severe symptomatology by different viruses and SARS-CoV-2. As mentioned, RNA viruses need the active NF-κB pathway within host cells to replicate, and SARS-CoV-2 is no exception; therefore, drugs that inhibit NF-κB activation potentially reduce viral replication. NAC has been shown to inhibit the NF-κB pathway as well as replication of human influenza viruses (H5N1, Vietnam/VN1203 strain) in human lung epithelial cells in a dose-dependent manner (5 to 15 mM) [34,42,43]. Besides, NAC has also been shown to inhibit the replication of viruses such as human immunodeficiency virus (HIV) and respiratory syncytial virus (RSV) and SARS-CoV-2 [39,44]. In SARS-Cov-2, major protease (Mopar) is required for viral replication, and it has been suggested that NAC bind to Cys-145, an active Mopar site inhibiting its protease activity, interfering with virus entry into the cell and with viral replication. Therefore, NAC could serve as a first-line drug specifically for viral infections such as that caused by SARS-Cov-2 [33]. It has been shown that NAC can change the redox balance to a reduced status by replenishing reduced glutathione (GSH). It also suppresses NF-κB activation at concentrations of 10 mm or more, resulting in the modulation of cytokine and chemotherapeutic signal production, making NAC an important substance for treating viral infections, including SARS-CoV-2 [30,34,43,45].
The SARS-Cov-2 infection triggers ROS production, which induces the stabilization of hypoxia-inducible factor 1α (HIF-1α) and the action of this factor inhibits T-cell response reduces epithelial cell survival [46]. Similarly, SARS-CoV-2 infected cells adapt their metabolism after infection and become highly glycolytic, facilitating SARS-CoV-2 replication. Antioxidants administration, such as NAC, reduces ROS and decreases glycolysis [46]. This condition increases the severity of viral infections in this population. Increased cellular oxidation with aging and illness is the most likely explanation for the increased vulnerability of the elderly and people with comorbidities and underlying health problems to viral infections, including that caused by SARS-CoV-2. Similarly, all respiratory RNA viruses induce Nox2-mediated ROS production in alveolar macrophage endosomes. In turn, neutrophils and monocytes recruitment from the bloodstream to the infection site may contribute significantly to a collateral increase in ROS production during infection [6].
Alteration of viruses’ entry and foldingOur research group has reported that the cell-surface protein disulfide isomerase (PDI), rich in cysteine residues (Cys), interacts with rotavirus, and this interaction decreases when treated with agents that block thiol-disulfide exchange or alter cellular redox status (DTNB, Bacitracin, N-ethylmaleimide, and N-acetylcysteine) [5,8-11,47]. We found that due to thiol/disulfide exchange inhibitors, PDI interaction with rotavirus triple-layer particles (TLP) or with their structural proteins is reduced. The TLPs interactions with cell membrane enriched fractions of mouse villi produce reordering in the rotavirus structural proteins' disulfide bridges. This suggests that PDI interacts with rotavirus virions through redox reactions facilitating rotavirus entry into the host cell, supporting the idea that PDI interacts with rotavirus and participates in the redox mechanisms that rotavirus requires to enter the cell [48].
SARS-CoV-2 Spike (S) protein, as in many SARS-related coronaviruses, and the SARS cell receptor, angiotensin-2 converting enzyme (ACE2), are rich cysteine residues and form intramolecular disulfide bonds. From simulations of the SARS CoV-2 molecular dynamic, it has been observed that there is higher domain flexibility when the four disulfide bond domains are reduced in the binding of the RBD on the spike domain to the ACE2 receptor. This occurs especially in the 456-490 residue that interacts with the ECA2 cell receptor [49]. Disulfide bonds in ECA2 protein have potential redox activity, facilitating primary interaction between the receptor and the S protein. Infection-resistant animals lack the ability to oxidize or reduce by having mutated one of the cysteines in ECA2 sequences (Cys133-Cys141), supporting the redox hypothesis virus-ECA2 interaction. Additionally, ECA2 is a known regulator of oxidative stress; the overexpression of ECA2 prevents the angiotensin II-induced increase in ROS and NADPH oxidase expression in the endothelium [32,50]. NAC can block ECA2 activity by reducing disulfides, which would make it difficult for SARS-CoV-2 to penetrate target cells [50] a similar to how NAC interferes with the binding of rotavirus to membrane PDIs [48] because it interferes with the redox status.
Reports from our rotavirus research group suggest that PDI, and even other related thioredoxins or integrin αvβ3, are potential candidates for these reactions since the anti-PDI antibodies and thiol-disulfide exchange inhibitors can significantly reduce PDI-TLP interaction and also rotavirus infection [47].
It is unusual for viruses to use a single receptor molecule; they generally use several, located in different organs, which fulfill the same function: binding, conformational changes of viral particles, and entry of the virus into the cell. Our research group has proposed for rotavirus infection [5]: (I) Binding molecules represented by sialic acid and some integrins; (II) Accompanying molecules including Hsc70 and other heat shock proteins; (III) Molecules with redox function such as PDI, Erp57, and other related thioredoxins and (IV) Molecules involved in the endocytosis process or other alternate mechanisms, these last ones can become part of the events I-III.
The main limitation in the entrance of a virus to the host cell is to find the set of molecules that allow it to infect, especially when it is in the transit of colonizing a new species. In this case, a high viral load is required to infect individuals of non-susceptible species and low viral load when it acquires the mutations that allow it to reach, with high affinity, the receptors that facilitate the infection. Receptor molecules that execute the entry mechanism may differ partially or completely depending on the species, cell line, and viral strain [5]. The use of receptors and the virus's tropism would be determined by the relative abundance and physical proximity of the receptors on the host cell's surface. The virus's structural proteins, involved in the first steps of the infectious cycle, are substrates of the molecules on the cell surface with oxidoreductase, thiol isomerase, and chaperone activities responsible for the conformational changes that these viral interaction proteins need to ensure internalization.
Decrease in virions production per infected cellIntracellular oxidative stress contributes to the folding of viral proteins because these require oxidation to form sulfur bridges, a necessary event to assemble mature and infectious virions. RNA viruses induce oxidative stress, and by treating cells with antioxidants, a non-conducive environment for viral replication is generated. Therefore, the number of virions per infected cell decreases [8,43,51], giving the organism time to react and control it. Of all known antioxidants, the oldest and most studied is NAC, pharmacokinetics, and minimum, maximum, and toxic doses are known. The lethal dose 50, (LD50) oral (rat): 5050 mg/kg; DL50 oral (mouse): 4400 mg/kg; DL50 intraperitoneal (mouse): 400 mg/kg (see RTECS Number: HA1660000). Although no data are available for humans, up to 150 mg/kg have been reported for intravenous acetaminophen poisoning without adverse reactions. NAC's final infusion rate can be from 12.5 mg to 18.75 mg/kg/h depending on acetaminophen intake; or orally, with intakes of 32 g/day of NAC [52]. For this reason, NAC, together with many other compounds that activate the anti-inflammatory PPAR pathway (fibrates, vitamin A, DHA) and with recognized antioxidants such as vitamin D, vitamin E, vitamin C, and NSAIDs can be used in infected patients to decrease the number of virions produced per cell. The amounts used should be by symptomatology, according to medical criteria. Low amount when symptoms are mild, moderate when symptoms are medium, and high when symptoms are strong, or patients have comorbidities. If RNA viruses, including SARS-CoV-2, stimulate pro-inflammatory pathways such as NF-κB and oxidants such as ROS and inhibit anti-inflammatory ones such as PPAR, the ideal is to use drugs that also act on all three sites. It is unlikely that a single drug would inhibit the acute infection since RNA viruses, by various mechanisms, act on all three of these pathways. In our model with rotavirus, we found that the different drugs analyzed have a hierarchical order in the force of action to decrease the infective capacity, as follows: the most potent is NAC, then NF-κB inhibitors, PPAR agonists, and finally vitamins [9-11]. An ideal scheme for managing viruses, including SARS-CoV-2, would be to use drugs that act on all three events, having NAC as a base, along with the other compounds. This scheme seeks to keep virions generation low per cell and allow sustained production of viral antigens to develop an efficient and balanced immunity, similar to what is sought in the vaccination with the complete inactive or attenuated virus.
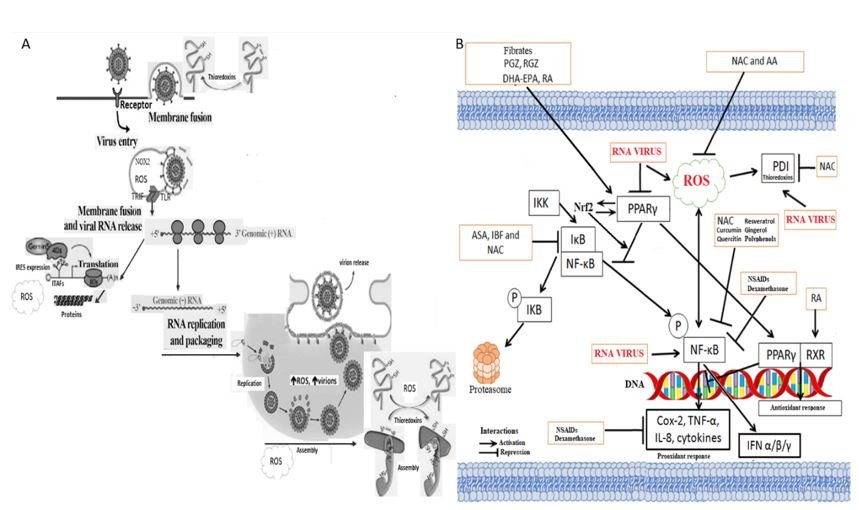
RNA viruses generate ROS, stimulate the inflammatory pathway NFkB and inhibit the anti-inflammatory pathway PPAR γ.
A. Activation of toll-like receptor 7 (TLR7) by viral RNA trapped within endosomes provides a key stimulus for the induction of type 1 interferon by RNA viruses. Such viruses evoke superoxide (H2O2) production by the NOX2-dependent biochemical complexes of NADPH oxidase; for this effect, the presence of TLR7 is required. TIR-domain-containing adapter-inducing interferon-β (TRIF) is an adapter in responding to activation of toll-like receptors (TLRs). Furthermore, the subsequent generation of hydrogen peroxide within these endosomes leads to oxidation of Cys98 in TLR7 that blocks this receptor's ability to transmit a signal that increases the production of type 1 interferon. Antioxidants capable of inhibiting NOX2, promoting the elimination of H2O2, or assisting in the restoration of the native structure of Cys98 in TLR7, may increase the TLR7-mediated induction of type 1 interferon and antiviral antibodies. On the other hand, when viruses "take over" the endoplasmic reticulum's biochemical machinery to translate their RNA or replicate it, oxidative stress is generated due to total or partial inhibition of cellular protein synthesis. Likewise, under conditions of cell stress, the machinery that reads the IRES of the viral RAN to translate it into proteins is expressed. Simultaneously, the expression of the pro-inflammatory pathway NFkB is induced, which induces greater production of reactive oxygen species. Also, RNA viruses inhibit the anti-inflammatory pathway PPARγ. Viruses use this oxidative environment because it favors the assembly of proteins or the conformation of viral proteins that require disulfuric bridges (s-s). The oxidative environment helps generate virions more quickly, leave the cell, and infect other cells or individuals.
B.RNA viruses, including SARS-CoV-2, stimulate pro-inflammatory pathways such as NF-κB and oxidants such as ROS and inhibit anti-inflammatory ones such as PPAR; the ideal is to use drugs that also act on all three sites. Giving antioxidants such as N-acetyl cysteine (NAC) or others counteracts the oxidative environment by being H+ donors to neutralize H2O2, forming water. Similarly, antioxidants induce several peroxidase enzymes (they modify H2O2) and promote glutathione synthesis, a cofactor for specific peroxidases and a catalyst in reactions that reconvert oxidized cysteine groups to their native or reduced form. NAC is both a direct and indirect inhibitor of the NFkB pathway. This is important because most viruses (including RNAs and some DNAs) induce this pathway to generate reactive oxygen species, inducing the oxidative environment that favors virion production. NAC as a reducing agent reduces the intracellular oxidative environment, decreasing the production of virions, enabling a better immune response of the patient against the virus. A bidirectional loop exists between PPARγ and Nrf2.
In the cell membrane, RNA viruses require redox molecules that allow them to make the conformational changes in viral proteins necessary for binding and penetrating the cell. They need to induce cellular oxidative stress because generating cellular stress, the biochemical machinery necessary for the translation of viral RNA is expressed using IRES. Also, pro-oxidant cytokines such as tumor necrosis factor and other pro-inflammatory cytokines are generated. RNA viruses inhibit antioxidant enzymes such as superoxide dismutase, important factors in anti-inflammatory pathways such as Nrf2, PPARγ, etc. ROS stimulates the activation of the NF-κB pathway, and both increase viral replication. Treatment and use of antioxidants that act on all three pathways are potential therapeutic agents in viral RNA diseases, including SARS-CoV-2. Their use, both in animals and in human patients, leads to a decrease in the generation of virions per cell, allowing the sustained production of viral antigens to develop an efficient and balanced immunity, similar to what is sought in vaccination inactivated or attenuated the whole virus.
Carlos A Guerrero declare no potential conflict of interest.
Copyright: © 2021 Carlos A Guerrero. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.








 Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Open Access by
Acta Scientific is licensed under a Creative Commons Attribution 4.0 International License
Based on a work at https://actascientific.com
ff